the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Feb 2025
| 27 Feb 2025
Base synergy in freshly nucleated particles
Galib Hasan
Yosef Knattrup
Sulfuric acid (SA), ammonia (AM), and dimethylamine (DMA) are believed to be key contributors to new particle formation (NPF) in the atmosphere. NPF happens through gas-to-particle transformation via cluster formation. However, it is not obvious how small clusters grow to larger sizes and eventually form stable aerosol particles. Recent experimental measurements showed that the presence of mixtures of bases enhanced the nucleation rate by several orders of magnitude.
Using quantum chemistry methods, this study explores this base synergy in the formation of large clusters from a mixture of SA, AM, and DMA. We calculated the binding free energies of the (SA)n(AM)x(DMA)n−x clusters, with n from 1 to 10, where x runs from 0 to n. The cluster structures were obtained using our recently developed comprehensive configurational sampling approach based on multiple ABCluster runs and meta-dynamic sampling via the Conformer–Rotamer Ensemble Sampling Tool (CREST). The structures and thermochemical parameters are calculated at the B97-3c level of theory. The final single point energy of the clusters is calculated at the ωB97X-D3BJ/6-311++G(3df,3pd) level of theory.
Based on the calculated thermochemistry, we found that AM, despite being a weaker base, forms more intermolecular interactions than DMA and easily becomes embedded in the cluster core. This leads to the mixed SA–AM–DMA clusters being lower in free energy compared to the pure SA–AM and SA–DMA clusters. We find that the strong base DMA is important in the very first steps in cluster formation, but for larger clusters an increased ammonia content is found. We also observed that the cluster-to-particle transition point for the mixed SA–AM–DMA clusters occurs at a cluster size of 14 monomers, which is notably smaller than the transition points for the pure SA–AM (16 monomers) or pure SA–DMA (20 monomers) systems. This indicates a strong synergistic effect when both AM and DMA are present, leading to the formation of stable freshly nucleated particles (FNPs) at smaller cluster sizes. These findings emphasize the importance of considering several base molecules when studying the formation and growth of FNPs.
- Article
(2048 KB) - Full-text XML
-
Supplement
(929 KB) - BibTeX
- EndNote




Atmospheric aerosols, particularly fine particles (< 1 µm in diameter) and ultrafine particles (< 100 nm in diameter), significantly impact human health by being the primary contributors to air-pollution-related mortality (Pelucchi et al., 2009; Cromar K, 2023). Aerosols also directly influence Earth's energy budget by scattering and absorbing solar radiation, resulting in cooling and warming effects, respectively (Loeb and Kato, 2002). Additionally, they have an even higher indirect effect on the global climate by serving as cloud condensation nuclei (CCN) for the formation of clouds, fog, or mist (Rosenfeld et al., 2014). To date, the interactions between aerosol particles and clouds remain the least understood processes in global climate estimation (Cooley et al., 2023).
Primary aerosols are directly emitted into the atmosphere, while secondary aerosols are formed through gas-to-particle conversion, resulting in the formation of freshly nucleated particles (FNPs). The formation of aerosols is initiated by forming strong hydrogen-bonded molecular clusters from different atmospheric vapour molecules (Kulmala et al., 2013). Clusters that possess strong intermolecular interactions can be stable against evaporation and further grow into aerosol particles of roughly 2 nm and above. Inorganic acids, such as sulfuric acid, and bases, such as ammonia and amines, are key components in the initial cluster formation in the atmosphere (Spracklen et al., 2006; Sipilä et al., 2010; Kirkby et al., 2011; Almeida et al., 2013). In addition, other chemical constituents are believed to influence clustering, such as ions originating from galactic cosmic rays (Kirkby et al., 2016) and condensation of highly oxygenated molecules (HOMs) (Bianchi et al., 2016). To understand the initial stages of atmospheric aerosol formation, it is essential to know the concentrations and chemical compositions of the clusters along with the gaseous compounds that contribute to their growth.
Measurements of clusters below 2 nm are extremely challenging, and no comprehensive, simultaneous field measurements of these clusters and their precursors have been conducted until now. Standard condensation particle counters (CPCs) typically have detection thresholds of around 2–3 nm, making them inadequate for detecting the smallest clusters (McMurry, 2000). While particle size magnifiers (PSMs) can detect clusters as small as ∼ 1.5 nm, they are expensive, have poor counting efficiency, and do not provide information about the chemical composition of the clusters (Vanhanen et al., 2011). Tools such as a chemical ionization atmospheric pressure interface time-of-flight (CI-APi-TOF) mass spectrometer are necessary for determining the chemical composition of growing clusters (Jokinen et al., 2012). However, these techniques alter the clusters' composition due to fragmentation during the measurement process, leading to potentially inaccurate results (Zapadinsky et al., 2018; Passananti et al., 2019; Alfaouri et al., 2022). The CI-APi-TOF can usually only detect clusters up to a certain size of around 10 acid molecules and 10 base molecules (Almeida et al., 2013). This leaves a knowledge gap in the chemical composition of large clusters in the range of 1.0 to 2.0 nm. As this is the size range for cluster stabilization, it is crucial to get a better understanding of this unknown cluster-to-particle transition regime (Kulmala et al., 2013). Wu et al. (2023) recently presented a robust computational framework that can be applied to study clustering from single molecules all the way up to 2 nm clusters. Such an approach offers an improved understanding of the chemical interactions and stability of these atmospheric clusters.
Ammonia (AM) and dimethylamine (DMA) are key contributors to the initiation of sulfuric acid (SA) nucleation and greatly enhance the particle formation rates compared to the pure sulfuric acid or sulfuric acid–water systems (Sipilä et al., 2010; Almeida et al., 2013). This happens due to the proton transfer reactions between the acid and base molecules, which leads to salt formation. Numerous quantum chemical studies have corroborated the role of bases such as AM (Ianni and Bandy, 1999; Larson et al., 1999; Nadykto and Yu, 2007; Kurtén et al., 2007; Loukonen et al., 2010; Herb et al., 2011; DePalma et al., 2012, 2014) and DMA (Kurtén et al., 2008; Loukonen et al., 2010; Kupiainen-Määttä et al., 2012; Ortega et al., 2012; Olenius et al., 2013b; Nadykto et al., 2014; Henschel et al., 2014; DePalma et al., 2012, 2014; Henschel et al., 2016; Ma et al., 2016) in stabilizing the initial SA clusters.
Recent experimental studies have demonstrated that the simultaneous inclusion of both ammonia and amines with sulfuric acid increases new particle formation rates by 10–100 times compared to mixtures containing only sulfuric acid and amines (Glasoe et al., 2015; Yu et al., 2012). This effect cannot be explained by the aqueous-phase base constant or the gas-phase proton affinity, suggesting that the underlying reason for this base synergy is not well understood. One possible explanation is that atmospheric ammonia concentrations are usually much higher than those of amines. However, laboratory experiments demonstrated that, when ammonia levels were lower than dimethylamine, the reactions between sulfuric acid and bases still produced nanoparticles with a higher ammonia content compared to dimethylamine (Lawler et al., 2016). This observation suggests that ammonia uptake is driven by a physicochemical effect in the early stages of particle formation rather than by the relative concentrations of the substances.
Temelso et al. (2018) provided theoretical evidence of base synergy by calculating free energies of three-component molecular clusters composed of sulfuric acid, amine (dimethylamine or trimethylamine), and ammonia. Their findings indicated that adding sulfuric acid to a cluster containing these mixed bases is thermodynamically more favourable than adding it to a cluster with only sulfuric acid and a single type of amine. Myllys et al. (2019) extended the work to larger clusters and provided a molecular-level explanation for the synergistic effects in SA–AM–DMA cluster formation, showing that ammonia can act as a “bridge former” and is more likely to be protonated than dimethylamine despite having a lower gas-phase basicity. Their quantum chemical simulations indicated that ammonia's inclusion can increase the particle formation rate by up to 5 orders of magnitude compared to the SA–DMA system. However, these studies have been limited to very small clusters of up to eight monomers and therefore do not give insight into the cluster-to-particle transition point of the mixed SA–AM–DMA clusters or the AM : DMA ratio in the growing clusters.
Previously, our group pushed the boundaries of studying large (SA)n(AM)n clusters, investigating systems with up to 60 molecules (n=30) in order to understand the transition from clusters to particles (Engsvang and Elm, 2022; Engsvang et al., 2023). The exponential increase in the number of possible configurations with respect to cluster size required an improved configurational sampling approach as described by Wu et al. (2023). In this work we extend our previous efforts in studying large clusters and perform quantum chemical (QC) calculations on mixed (SA)n(AM)x(DMA)n−x clusters, with n from 1 to 10 and . Hence, we study clusters with an acid : base ratio of 1 : 1 and all combinations of AM and DMA for each cluster size. We recently proposed a property-based criterion for defining FNPs as the boundary between discrete cluster configurations and bulk particles (Wu et al., 2024). Specifically, we defined FNPs as instances where one or more ions are fully embedded inside the cluster and where the change in the size-averaged binding free energy approaches zero. Hence, the emergence of FNPs acts as the cluster-to-particle transition point. Here we extend this concept to clusters with mixed bases. Our study suggests that mixed clusters possess varying stability, with the introduction of AM becoming increasingly favourable as the cluster size grows, facilitating the cluster-to-particle transition process and leading to FNPs at an earlier stage.
2.1 Computational details
Density functional theory (DFT) calculations during the configurational sampling procedures (single point energies, geometry optimization, and vibrational frequency calculations) were performed using the empirically corrected B97-3c method (Brandenburg et al., 2018) in the ORCA 5.0.4 quantum chemistry program (Neese, 2022). Single point energies using ωB97X-D3BJ (Najibi and Goerigk, 2018) with 6-311++G(3df,3dp) (Ditchfield et al., 1971), ma-def2-QZVPP (Zheng et al., 2011), ma-def2-TZVP (Zheng et al., 2011), and def2-TZVDP (Weigend and Ahlrichs, 2005) were also performed in ORCA 5.0.4. The GFN1-xTB (Grimme et al., 2017) and reparameterized GFN1-xTBre-par semi-empirical calculations (Wu et al., 2024) were performed using the xTB 6.4.0 program (Bannwarth et al., 2021). The reparameterization was performed according to the workflow given by Knattrup et al. (2024), where the energy and gradients of GFN1-xTB were optimized to fit the FNP structures and energies at the B97-3c level of theory. We switched to the GFN1-xTBre-par method once it became available. Hence, the overall sampling was performed with a mix of GFN1-xTB and GFN1-xTBre-par. The meta-dynamic simulations were performed using CREST in non-covalent interaction mode (Pracht et al., 2017, 2020; Pracht and Grimme, 2021; Grimme, 2019; Spicher et al., 2022). Initial clusters were generated with ABCluster version 3.2 (Zhang and Dolg, 2015, 2016) with a CHARMM force field (Huang and MacKerell, 2013).
2.2 Configurational sampling workflow
We study the (SA)n(AM)x(DMA)n−x clusters, with n from 1 to 10 and . This leads to n+1 compositions for each cluster size n, implying that we have to sample many of the largest (SA)7–10(base)7–10 cluster structures. We employed our recently established configurational sampling protocol presented by Wu et al. (2023, 2024), demonstrating an excellent balance between accuracy and computational cost. The configurational sampling procedure can be outlined as follows:
The initial cluster structures were generated through 10 parallel ABCluster runs, yielding a total of 10 000 local-minimum configurations. Our previous studies (Wu et al., 2023, 2024) confirmed that conducting multiple parallel ABCluster explorations provides more accurate predictions for the global energy minimum structures of large clusters compared to a single, prolonged exploration. We used ionic monomers but kept the overall cluster charge neutral in order to facilitate proton transfer, as this is typically observed in the lowest free energy clusters. Subsequent geometry optimizations were performed on all these clusters using the GFN1-xTB semi-empirical method (Grimme et al., 2017). DFT single point calculations were then performed using the B97-3c method (Brandenburg et al., 2018) on top of the GFN1-xTB-optimized conformers.
Next, we filtered out high-energy configurations by selecting only the 1000 lowest structures for partial optimization at the B97-3c level. This partial optimization step was chosen to save computational time and eliminate energetically high-lying configurations. From these, we chose the 100 lowest energy configurations for full optimization and vibrational frequency calculations.
The lowest free energy conformer was then selected for CREST exploration as suggested by Knattrup et al. (2024), which involves meta-dynamics to provide reasonably good geometries using the following workflow:
The CREST exploration was performed using GFN1-xTB/GFN1-xTBre-par in non-covalent interaction mode. We selected the 100 best geometries from the CREST optimization and performed full optimizations and quasi-harmonic vibrational frequency calculations (Grimme, 2012) to obtain the corresponding Gibbs free energies. The overall workflow is highly computationally demanding but should yield a good estimate of the lowest free energy structures.
2.3 Cluster binding free energies
We calculated the cluster standard binding free energies by subtracting the free energy of the cluster from the sum of the free energies of the individual monomers. This is calculated as follows:
In a similar manner, the electronic binding energies and the binding thermal correction of the free energy can be calculated. This allows the division of the binding free energy in the following terms:
Here we calculated the structures and thermochemistry at the B97-3c level (the ΔGbind,thermal term) and the binding electronic energy at the ωB97X-D3BJ/6-311++G(3df,3pd) level (the ΔEbind term). We refer to benchmark calculations in the Supplement for a justification for applying the ωB97X-D3BJ/6-311++G(3df,3pd) level for single point energies. It should be noted that the ΔGbind,thermal term is calculated using the quasi-harmonic approximation (Grimme, 2012) as a default in ORCA. The quasi-harmonic approximation removes spurious low vibrational modes but does not take local and global anharmonicity into account. The recent work by Halonen (2024) provides a promising avenue to further improve our understanding of cluster stability by deriving an analytical expression to account for local anharmonicity. Integrating this approach into future studies would increase the accuracy of our thermodynamic predictions, but as it is not straightforward to obtain the energy barriers between cluster configurations, this is beyond the scope of the current work.
The equations above only account for the thermochemistry of the clusters. To calculate the “actual” binding free energies under specific conditions, we use the self-consistent distribution function (Wilemski and Wyslouzil, 1995; Halonen, 2022):
Here pref corresponds to a reference pressure (1 atm) and pi represents monomer partial pressures. The self-consistent formulation allows us to correctly establish the monomer free energies as zero. We previously (Wu et al., 2024) tested various formulations of the actual free energies under given conditions and found no deviations between the calculated free energies.
2.4 Size-averaged binding free energies
From the standard binding free energies, we also calculate the size-averaged binding free energies () of the clusters. The physical interpretation of this quantity can be seen by analysing as a function of cluster size. As the cluster size increases, a convergence in is seen toward the formation of free energy in the bulk system (Sindel et al., 2022). The physical significance of can be realized by considering the difference in the average binding free energy between a very large (SA)99(base)99 cluster and a (SA)100(base)100 cluster. The addition of one extra acid–base pair would have little impact on the total free energy of the cluster, and therefore the gradient of becomes zero, resembling the bulk particle phase.
2.5 The convex hull approach
The emergence of fully coordinated ions inside the cluster yields information about the transition from discrete cluster configurations to the particle phase. In a small cluster, all monomers are fully exposed to the exterior, and a large stabilization in free energy is gained when adding more monomers to the cluster. In larger cluster structures, fully coordinated ions emerge, corresponding to a “solvated” ion with a solvation shell. Adding more monomers to the existing solvation shell leads to less stabilization free energy gained compared to a smaller cluster.
To investigate when the first fully coordinated ion appears in our calculated cluster structures, we applied the 3D convex hull approach as described by Wu et al. (2024). The applied algorithm is freely available at https://gitlab.com/AndreasBuchgraitz/clusteranalysis (last access: 12 February 2025).
2.6 Cluster-to-particle transition point
Sections 2.4 and 2.5 both yield inferred evidence of the transition from clusters to particles. Using the 3D convex hull approach, we can identify when the first solvation shell is formed in the clusters. Structurally, this implies that we are transitioning from a cluster towards a particle. Thermodynamically, when the size-averaged as a function of cluster size becomes constant (the gradient of the change in approaches zero), the cluster behaves more like the bulk than a cluster. Hence, we define the cluster-to-particle transition point as the point where both of these conditions are satisfied. That is, structurally, there must be development of a new phase by having at least one fully coordinated ion, and the thermochemistry must resemble the bulk by leveling out in as a function of cluster size. Putting a strict number on when the change in resembles the bulk is tricky and most likely system-dependent. Hence, tentatively, we assign a change of roughly ∼ 1 kcal mol−1 in the from cluster sizes m to m+1 as the convergence point.
3.1 Binding free energies under standard conditions
Applying the outlined extensive cluster sampling approach, we studied the mixed (SA)n(AM)x(DMA)n−x clusters, with n from 1 to 10 and . The pure SA–AM and SA–DMA clusters are taken from Wu et al. (2024), with refined ωB97X-D3BJ/6-311++G(3df,3pd) single point energies calculated in this work. Figure 1a presents the standard binding free energies, calculated at 298.15 K and 1 atm, as a function of the number of monomers m in the cluster. Each point on the graph is labeled with a pair (AM, DMA) indicating the numbers of ammonia and dimethylamine monomers in the cluster.
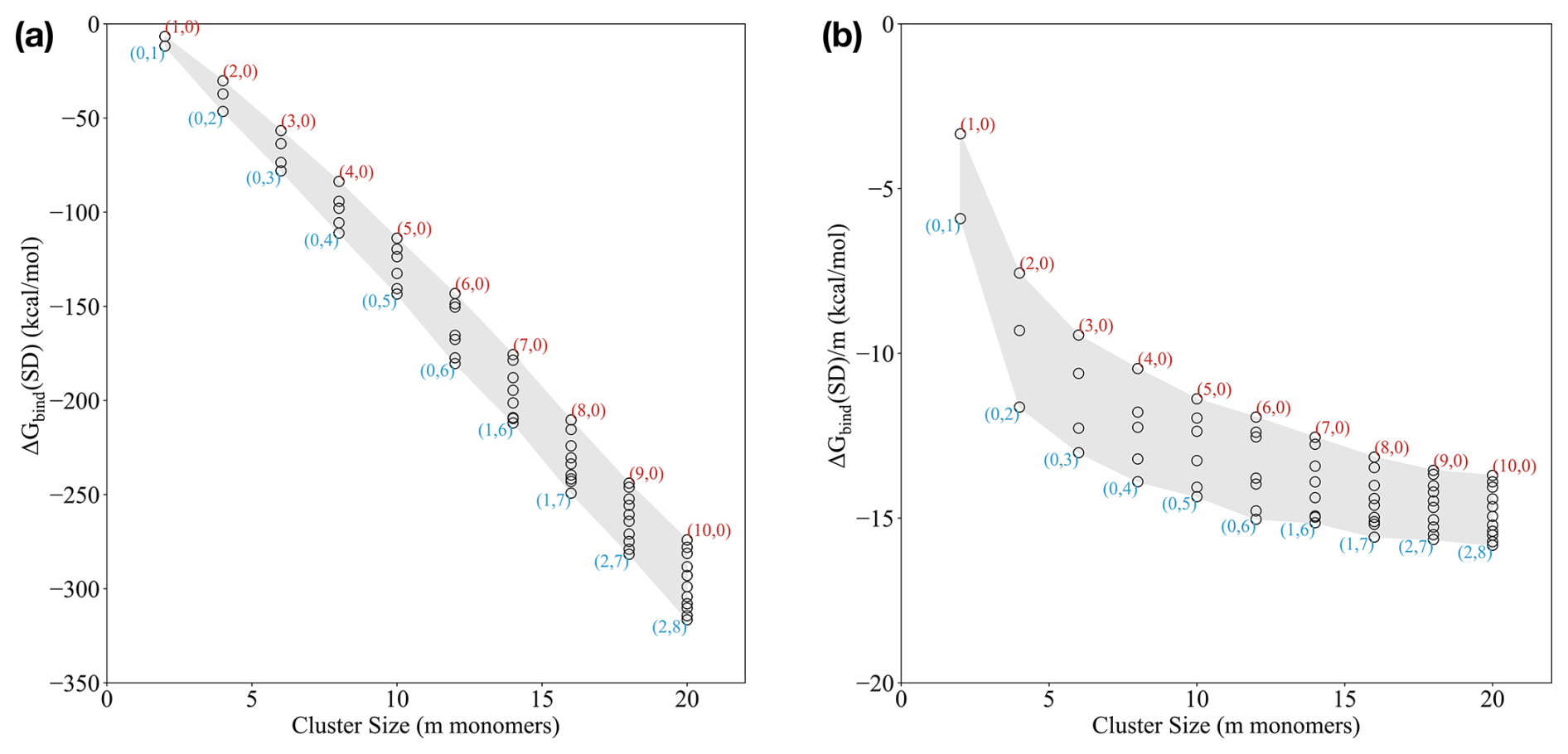
Figure 1(a) Binding Gibbs free energy and (b) size-averaged binding Gibbs free energy of the (SA)n(AM)x(DMA)n−x clusters, with n from 1 to 10 and under standard conditions (298.15 K and 1 atm). The free energies are calculated at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
Based on the standard binding free energies (Fig. 1a), we see that the SA–DMA clusters are the most stable ones up to 12 monomers. Beyond this point, it is more favourable to exchange one to two DMA molecules with AM. Comparing the AM : DMA ratio for smaller clusters, the (5,0) cluster is significantly higher in free energy, by almost 20 kcal mol−1, compared to the (0,5) cluster, indicating that DMA alone provides substantial stability for the small cluster sizes.
For cluster sizes of around 14–16 monomers, compositions only containing SA and DMA are correspondingly 2.9 and 9.4 kcal mol−1 higher in free energy than (1,6) and (1,7), suggesting that introducing one AM molecule increases the stability compared to only having DMA in the clusters. This trend is also observed for larger clusters, where the clusters with compositions only containing SA and DMA are higher in free energy compared to (2,7) and (2,8), indicating that having one or two AM molecules in addition to DMA provides higher stability for the larger clusters.
Figure 1b presents the size-averaged binding free energies (). These values represent the average binding free energy of each molecule in the cluster. Similar to our previous work (Engsvang and Elm, 2022; Engsvang et al., 2023; Wu et al., 2023, 2024), we see that the size-averaged free energy rapidly decreases as a function of cluster size and levels out around 12–20 monomers. This can be interpreted as the cluster transitioning towards more particle-like properties.
Using the convex hull method of Wu et al. (2024), we studied the formation of solvation shells for clusters with the lowest free energy at each size. When the cluster is only composed of SA and DMA (6,0), no encapsulation of ions occurs. However, when AM is added, the lowest free energy structure favours a shell structure, with AM encapsulated by SA and DMA in clusters of sizes (1,6) to (1,7) (see Fig. 2a). When two AM molecules are introduced, a single solvation shell forms, encapsulating both AM monomers as shown in Fig. 2b.
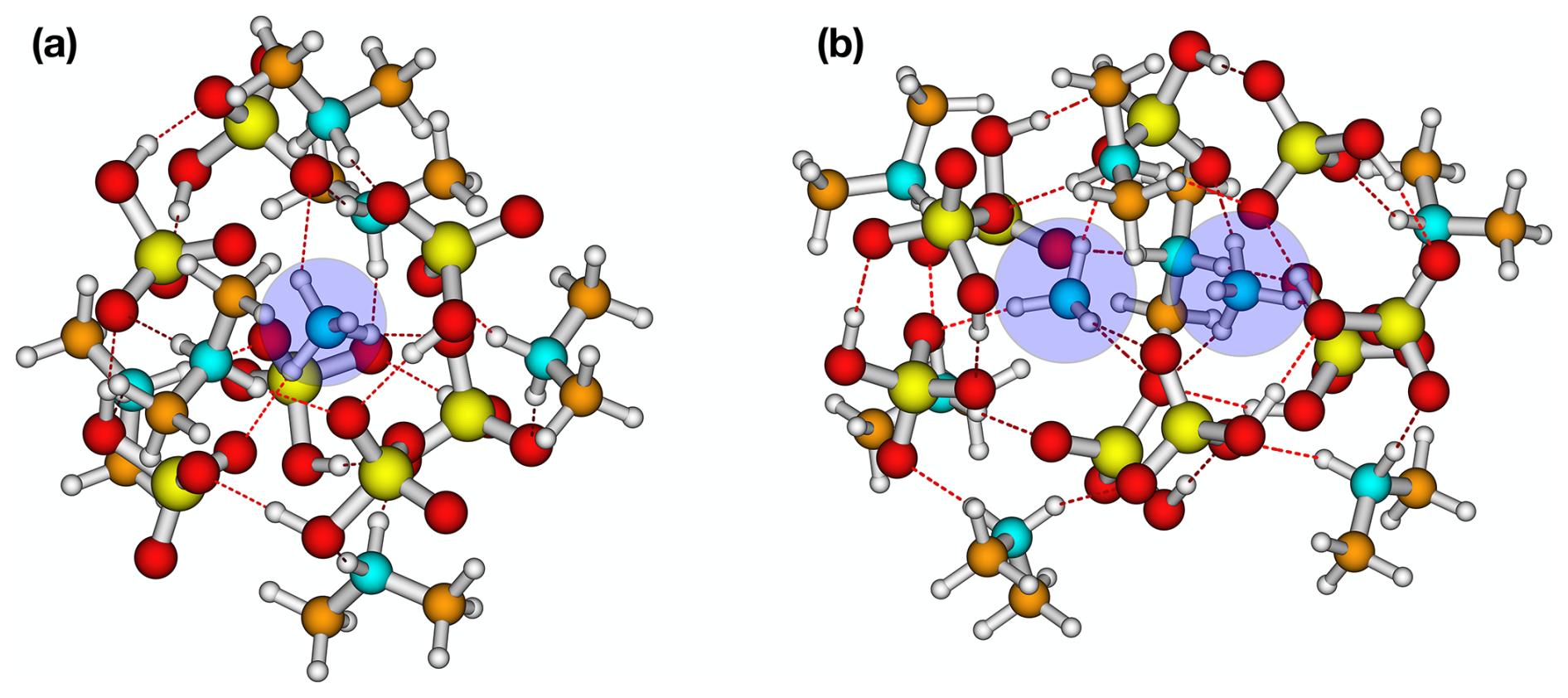
Figure 2Two examples of protonated ammonia (blue circles) encapsulated for (a) (SA)7(AM)1(DMA)6 and (b) (SA)9(AM)2(DMA)7. The structures are lowest in free energy at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory. White: hydrogen, blue: nitrogen, yellow: sulfur, red: oxygen, and brown: carbon.
The reason for the more stable clusters containing one to two AM molecules could be an intricate combination of hydrogen bond capacity, base strength, and steric hindrance. For instance, the Td symmetry of the AM molecule makes it capable of forming four intermolecular bonds, whereas DMA can only form two intermolecular bonds. This will become increasingly important as the cluster becomes larger and thus more spherical, as it increases possible coordination. In addition, the presence of the two bulky methyl groups in DMA imposes a strong steric hindrance. As a result, the protonated AM stays embedded at the centre of the cluster and forms a fully coordinated complex with the surrounding ions. This trend suggests that AM starts to play a more significant role in stabilizing larger clusters, and its presence becomes increasingly favourable as the cluster size grows. This is consistent with the experimental work by Lawler et al. (2016) that found increased AM content compared to DMA in newly formed SA–AM–DMA nanoparticles. A consequence of these findings is that the cluster-to-particle transition point differs significantly in the mixed-base clusters compared to the pure SA–AM and SA–DMA clusters. Hence, the cluster-to-particle transition point occurs at a cluster size of 14 monomers for SA–AM–DMA, compared to 16 for the SA–AM system and 20 for the SA–DMA system. These FNP sizes are based on us seeing the first emergence of a fully coordinated ion and a levelling out of the size-averaged free energies (see the definition in Sect. 2.6). In addition, these cluster-to-particle transition points correspond to 15.0, 16.7, and 17.3 Å for the SA–AM–DMA, SA–AM, and SA–DMA systems, respectively. We note that the cluster-to-particle transition point identified from our previous work (Wu et al., 2024) was unchanged by the single point refinement. Overall, this implies that there is a synergistic effect between the bases AM and DMA for the formation of FNPs.
3.2 Binding free energies under given conditions
Based on the calculated binding Gibbs free energies under standard conditions described in the previous section, we can evaluate the binding free energies under certain monomer concentration and temperature conditions using Eq. (3). Figure 3 presents the binding free energies of the clusters at 278.15 and 298.15 K. We considered two specific conditions: a low-concentration regime ([SA] = 107 molec. cm−3, [DMA] = 1 ppt, [AM] = 10 ppt) and a high-concentration regime ([SA] = 107 molec. cm−3, [DMA] = 10 ppt, [AM] = 10 ppb). These concentrations and temperatures align with typical conditions employed in the CLOUD chamber experiments (Almeida et al., 2013; Kürten et al., 2018) and real-world nucleation observations (Kürten et al., 2014). To better relate our findings to real atmospheric environments, our studied temperature and concentration regimes can be linked to conditions observed in the real atmosphere. For instance, in line with previous studies conducted in Hyytiälä, Finland, which represent typical boreal forest environments, we considered SA concentrations and temperatures that are relevant for new particle formation (NPF) processes. In these environments, SA concentrations are often in the range [SA] = 104–108 molec. cm−3, with temperatures that generally range between 278.15 and 298.15 K, depending on seasonal variations. These temperature and concentration conditions closely resemble the boundary layer conditions that are frequently encountered in temperate regions (Jokinen et al., 2022).
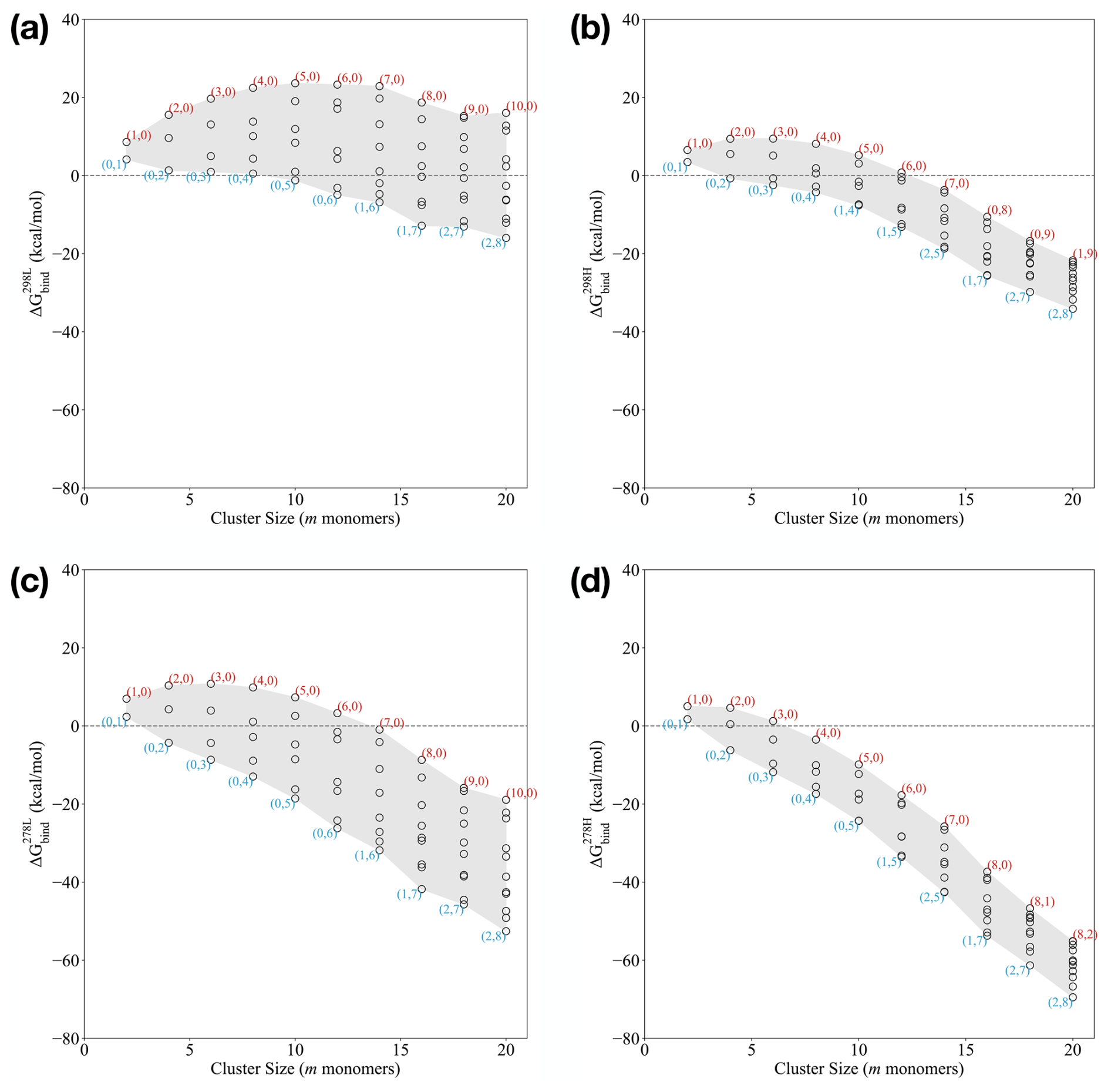
Figure 3Binding Gibbs free energy at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory of the (SA)n(AM)x(DMA)n−x clusters, with n=x between 1 and 10. The monomer concentration of [SA] was fixed at 107 molec. cm−3. “High conc.” refers to [AM] = 10 ppb and [MA] = [DMA] = 10 ppt. “Low conc.” refers to [AM] = 10 ppt and [MA] = [DMA] = 1 ppt. (a) 298.15 K: low conc., (b) 298.15 K: high conc., (c) 278.15 K: low conc., and (d) 278.15 K: high conc.
However, we do note that, for clean environments, the “low conc.” regime applied here might still represent an upper bound. We also tested the effect of decreasing [SA] to 106 molec. cm−3 or increasing it to 108 molec. cm−3 (see Figs. S1–S3 in the Supplement). In the following, we will go through each scenario.
3.2.1 Low concentrations at 298.15 K
Similar to the standard free energies in the previous section, the actual free energies in Fig. 3a show that the clusters have lower free energy when they are composed of one to two AM molecules for the larger cluster sizes. This scenario is seen from the smallest composition with 14 monomers to the largest cluster composition with 20 monomers. However, the clusters do predominantly contain DMA compared to AM. Across all the studied cluster sizes, the clusters with a high AM content are generally higher in binding free energies compared to those with a mix of AM and DMA or a higher DMA content. We see that the pure SA–AM clusters have a nucleation barrier with a critical cluster size at six monomers. This is not the case when the clusters are only composed of SA–DMA and is less pronounced for the SA–AM–DMA clusters with mixed bases. These findings align with the experimental results of Glasoe et al. (2015), which demonstrated that the formation of 1.8 nm sulfuric acid–base particles followed the trend AM < MA < DMA < TMA, where MA is methylamine and TMA is trimethylamine.
At a low base concentration, changing the SA concentration has a major influence on the results (see the Supplement). At [SA] = 106 molec. cm−3 there is a nucleation barrier in all the systems, and particles are unlikely to form. At [SA] = 108 molec. cm−3 the critical cluster is reduced to four monomers.
3.2.2 High concentrations at 298.15 K
In Fig. 3b, we see a much narrower span in the binding free energies under given conditions compared to Fig. 3a. This is primarily caused by increased stability of the SA–AM clusters due to a higher concentration of AM. By contrast, the SA–DMA clusters are much less affected by the increased concentration as they already have relatively low evaporation rates. The binding free energies are generally more negative compared to the low-concentration regime at 298.15 K, logically indicating greater stability at higher concentrations. These results are consistent with the previous work by Olenius et al. (2013a), Besel et al. (2020), and Kubecka et al. (2023). Due to the high concentration of AM = 10 ppb, we see AM molecules emerging in the lowest free energy clusters at much smaller sizes, e.g. (1,4) and (1,5). In a similar manner, two AM molecules are also found in the cluster at a smaller size of (2,5).
Similar to the low-concentration scenario, lowering the SA concentration to 106 molec. cm−3 leads to higher free energies (see the Supplement). Interestingly, this also leads to the emergence of three AM molecules in the m=16 cluster. Increasing the SA concentration to 108 molec. cm−3 does not change the trends of the system.
3.2.3 Low concentrations at 278.15 K
Looking at the binding free energies at 278.15 K and the low concentrations (Fig. 3c), the clusters with one to two ammonia molecules are again lowest in free energies at larger sizes. This scenario is observed across all the cluster compositions. Clusters such as (1,6–7) and (2,7–8) show the lowest binding free energies, indicating that the clusters are more stable with a higher proportion of DMA. Hence, the compositions of the lowest free energy clusters are consistent with the 298.15 K and low-concentration systems shown in Fig. 3a. This could indicate that the concentration and not the temperature is the primary driver in determining the lowest free energy cluster compositions. We see a small nucleation barrier in the SA–AM system with a critical cluster of six monomers but no barrier in the other systems.
Changing the SA concentration does not change the trends at 278.15 K and the low base concentration (see the Supplement).
3.2.4 High concentrations at 278.15 K
Figure 3d shows similar trends to the situation at 298.15 K as well as high concentrations (Fig. 3b). Hence, the lowest free energy composition is the same as at the higher temperature. Obviously, the clusters are lower in free energy compared to the higher temperature. No nucleation barriers are seen in any of the studied systems, with the free energy surface being downhill. Again, at low temperature, changing the SA concentration does not change the trends (see the Supplement).
Overall, our results show that, at low concentrations, the inclusion of DMA in the clusters tends to yield lower and more negative binding free energies. In contrast, clusters with a higher proportion of AM alone are less stable in the low-concentration regime and more stable in the high-concentration regime. These findings underscore the importance of DMA in the initial cluster stabilization regime but also show the importance of ammonia for facilitating the cluster-to-particle transition, leading to the onset of FNPs in the FNP regime. These results align with the previous hypothesis by Elm et al. (2017) and Elm (2017, 2020) that suggested that strong bases like DMA or diamines play a crucial role in the very initial stages of cluster formation, while the subsequent growth is driven by weaker bases such as AM.
Here we studied the formation of large clusters composed of sulfuric acid (SA), ammonia (AM), and dimethylamine (DMA). Using quantum chemical methods, we studied the mixed (SA)n(AM)x(DMA)n−x cluster systems, with n from 1 to 10 and at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory. We found that the pure SA–DMA clusters are the most stable up to a cluster size of around 8–12 monomers, depending on precursor concentrations, without the need for AM. As the cluster size increases beyond 10–14 monomers, adding one to three ammonia molecules significantly increases the stability of the cluster. This suggests a synergistic effect where the presence of a small number of ammonia molecules, in addition to DMA, enhances the overall stability of the sulfuric acid clusters, especially at larger cluster sizes. Additionally, in most of the clusters, AM molecules are embedded in the core, creating strong intermolecular interactions with SA, while the DMA molecules reside on the periphery of the cluster. Moreover, we found that the cluster-to-particle transition point in the mixed SA–AM–DMA system occurs at a smaller cluster size of 14 monomers, in contrast to the 16 monomers for SA–AM and the 20 monomers for SA–DMA found in the previous study by Wu et al. (2024). This suggests a significant synergistic effect when both AM and DMA are present, resulting in the formation of freshly nucleated particles (FNPs) at smaller cluster sizes. The identified base synergy between AM and DMA indicates that nucleation mechanisms are inherently complex, and further work is require to study the synergistic effects between other vapours. Hence, additional vapours of methylamine (MA) and methane sulfonic acid (MSA) could be interesting to study in the future, together with the growth of FNPs via uptake of SA, bases, and organics.
The code used to compute the solvation shells with the convex hull algorithm is available at https://gitlab.com/AndreasBuchgraitz/clusteranalysis (Jensen, 2024).
All the calculated structures and the thermochemistry are available in the Atmospheric Cluster Database (ACDB) https://doi.org/10.1021/acsomega.9b00860 (Elm, 2019).
The supplement related to this article is available online at https://doi.org/10.5194/ar-3-101-2025-supplement.
Conceptualization: JE. Methodology: GH, HW, YK, and JE. Formal analysis: GH, HW, and YK. Investigation: GH, HW, and YK. Resources: JE. Writing – original draft: GH, HW, YK, and JE. Writing – review and editing: GH, HW, YK, and JE. Visualization: GH, HW, and YK. Project administration: JE. Funding acquisition: JE. Supervision: JE.
At least one of the (co-)authors is a member of the editorial board of Aerosol Research. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Research Council's Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The numerical results presented in this work were obtained at the Centre for Scientific Computing in Aarhus (https://phys.au.dk/forskning/faciliteter/cscaa/, last access: 12 February 2025).
This study was funded by the European Union (ERC and ExploreFNP; grant no. 101040353). This work was funded by the Danish National Research Foundation (grant no. DNRF172) through the Center of Excellence for Chemistry of Clouds.
This paper was edited by Jose Castillo and reviewed by two anonymous referees.
Alfaouri, D., Passananti, M., Zanca, T., Ahonen, L., Kangasluoma, J., Kubečka, J., Myllys, N., and Vehkamäki, H.: A study on the fragmentation of sulfuric acid and dimethylamine clusters inside an atmospheric pressure interface time-of-flight mass spectrometer, Atmos. Meas. Tech., 15, 11–19, https://doi.org/10.5194/amt-15-11-2022, 2022. a
Almeida, J., Schobesberger, S., Kürten, A., et al.: Molecular Understanding of Sulphuric Acid-Amine Particle Nucleation in the Atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013. a, b, c, d
Bannwarth, C., Caldeweyher, E., Ehlert, S., Hansen, A., Pracht, P., Seibert, J., Spicher, S., and Grimme, S.: Extended tight-binding quantum chemistry methods, WIREs Comput. Mol. Sci., 11, e1493, https://doi.org/10.1002/wcms.1493, 2021. a
Besel, V., Kubecka, J., Kurten, T., and Vehkamaki, H.: Impact of quantum chemistry parameter choices and cluster distribution model settings on modeled atmospheric particle formation rates, J. Phys. Chem. A, 124, 5931–5943, 2020. a
Bianchi, F., Tröstl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., Bukowiecki, N., Chen, X., Duplissy, J., Gysel, M., Hutterli, M., Kangasluoma, J., Kontkanen, J., Kürten, A., Manninen, H. E., Münch, S., Peräkylä, O., Petäjä, T., Rondo, L., Williamson, C., Weingartner, E., Curtius, J., Worsnop, D. R., Kulmala, M., Dommen, J., and Baltensperger, U.: New particle formation in the free troposphere: A question of chemistry and timing, Science, 352, 1109–1112, https://doi.org/10.1126/science.aad5456, 2016. a
Brandenburg, J. G., Bannwarth, C., Hansen, A., and Grimme, S.: B97-3c: A revised low-cost variant of the B97-D density functional method, J. Chem. Phys., 148, 064104, https://doi.org/10.1063/1.5012601, 2018. a, b
Cooley, S., Schoeman, D., Bopp, L., et al.: Oceans and Coastal Ecosystems and Their Services [Internet], Climate Change 2022 – Impacts, Adaptation and Vulnerability, Cambridge University Press; 379–550 pp., https://doi.org/10.1017/9781009325844.005, 2023. a
Cromar, K. and Lazrak, N.: Risk communication of ambient air pollution in the WHO European Region: review of air quality indexes and lessons learned, World Health Organization, Regional Office for Europe, https://iris.who.int/handle/10665/365787, License: CC BY-NC-SA 3.0 IGO, 2023. a
DePalma, J. W., Bzdek, B. R., Doren, D. J., and Johnston, M. V.: Structure and Energetics of Nanometer Size Clusters of Sulfuric Acid with Ammonia and Dimethylamine, J. Phys. Chem. A, 116, 1030–1040, 2012. a, b
DePalma, J. W., Doren, D. J., and Johnston, M. V.: Formation and Growth of Molecular Clusters Containing Sulfuric Acid, Water, Ammonia, and Dimethylamine, J. Phys. Chem. A, 118, 5464–5473, 2014. a, b
Ditchfield, R., Hehre, W. J., and Pople, J. A.: Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules, J. Chem. Phys., 54, 724–728, https://doi.org/10.1063/1.1674902, 1971. a
Elm, J.: Elucidating the limiting steps in sulfuric acid–base new particle formation, J. Phys. Chem. A, 121, 8288–8295, 2017. a
Elm, J.: An Atmospheric Cluster Database Consisting of Sulfuric Acid, Bases, Organics, and Water, ACS Omega, 4, 10965–10974, 2019. a
Elm, J.: Toward a holistic understanding of the formation and growth of atmospheric molecular clusters: a quantum machine learning perspective, J. Phys. Chem. A, 125, 895–902, 2020. a
Elm, J., Passananti, M., Kurtén, T., and Vehkamaki, H.: Diamines can initiate new particle formation in the atmosphere, J. Phys. Chem. A, 121, 6155–6164, 2017. a
Engsvang, M. and Elm, J.: Modeling the Binding Free Energy of Large Atmospheric Sulfuric Acid–Ammonia Clusters, ACS Omega, 7, 8077–8083, 2022. a, b
Engsvang, M., Kubečka, J., and Elm, J.: Toward Modeling the Growth of Large Atmospheric Sulfuric Acid–Ammonia Clusters, ACS Omega, 8, 34597–34609, 2023. a, b
Glasoe, W., Volz, K., Panta, B., Freshour, N., Bachman, R., Hanson, D., McMurry, P., and Jen, C.: Sulfuric acid nucleation: An experimental study of the effect of seven bases, J. Geophys. Res.-Atmos., 120, 1933–1950, 2015. a, b
Grimme, S.: Supramolecular Binding Thermodynamics by Dispersion-corrected Density Functional Theory, Chem. Eur. J., 18, 9955–9964, 2012. a, b
Grimme, S.: Exploration of chemical compound, conformer, and reaction space with meta-dynamics simulations based on tight-binding quantum chemical calculations, J. Chem. Theory Comput., 15, 2847–2862, 2019. a
Grimme, S., Bannwarth, C., and Shushkov, P.: A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z=1–86), J. Chem. Theory Comput., 13, 1989–2009, 2017. a, b
Halonen, R.: A consistent formation free energy definition for multicomponent clusters in quantum thermochemistry, J. Aerosol Sci., 162, 105974, https://doi.org/10.1016/j.jaerosci.2022.105974, 2022. a
Halonen, R.: Assessment of Anharmonicities in Clusters: Developing and Validating a Minimum-Information Partition Function, J. Chem. Theory Comput., 20, 4099–4114, https://doi.org/10.1021/acs.jctc.4c00121, 2024. a
Henschel, H., Navarro, J. C. A., Yli-Juuti, T., Kupiainen-Määttä, O., Olenius, T., Ortega, I. K., Clegg, S. L., Kurtén, T., Riipinen, I., and Vehkamäki, H.: Hydration of atmospherically relevant molecular clusters: Computational chemistry and classical thermodynamics, J. Phys. Chem. A., 118, 2599–2611, 2014. a
Henschel, H., Kurtén, T., and Vehkamäki, H.: Computational study on the effect of hydration on new particle formation in the sulfuric acid/ammonia and sulfuric acid/ dimethylamine systems, J. Phys. Chem. A., 120, 1886–1896, 2016. a
Herb, J., Nadykto, A. B., and Yu, F.: Large Ternary Hydrogen-bonded Pre-nucleation Clusters in the Earth's Atmosphere, Chem. Phys. Lett., 518, 7–14, 2011. a
Huang, J. and MacKerell Jr., A. D.: CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data, J. Comput. Chem., 34, 2135–2145, 2013. a
Ianni, J. C. and Bandy, A. R.: A Density Functional Theory Study of the Hydrates of NH3⋅H2SO4 and its Implications for the Formation of New Atmospheric Particles, J. Phys. Chem. A., 103, 2801–2811, 1999. a
Jensen, A. B.: ClusterAnalysis, Gitlab [code], https://gitlab.com/AndreasBuchgraitz/clusteranalysis (last access: 12 February 2025), 2024. a
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012. a
Jokinen, T., Lehtipalo, K., Thakur, R. C., Ylivinkka, I., Neitola, K., Sarnela, N., Laitinen, T., Kulmala, M., Petäjä, T., and Sipilä, M.: Measurement report: Long-term measurements of aerosol precursor concentrations in the Finnish subarctic boreal forest, Atmos. Chem. Phys., 22, 2237–2254, https://doi.org/10.5194/acp-22-2237-2022, 2022. a
Kirkby, J., Curtius, J., Almeida, J., et al.: Role of Sulphuric Acid, Ammonia and Galactic Cosmic Rays in Atmospheric Aerosol Nucleation, Nature, 476, 429–433, 2011. a
Kirkby, J., Duplissy, J., Sengupta, K., et al.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, 2016. a
Knattrup, Y., Kubečka, J., Wu, H., Jensen, F., and Elm, J.: Reparameterization of GFN1-xTB for Atmospheric Molecular Clusters: Applications to Multi-Acid–Multi-Base Systems, RSC Adv., 14, 20048–20055, https://doi.org/10.1039/D4RA03021D, 2024. a, b
Kubecka, J., Neefjes, I., Besel, V., Qiao, F., Xie, H.-B., and Elm, J.: Atmospheric sulfuric acid–multi-base new particle formation revealed through quantum chemistry enhanced by machine learning, J. Phys. Chem. A, 127, 2091–2103, 2023. a
Kulmala, M., Kontkanen, J., Junninen, H., et al.: Direct observations of atmospheric aerosol nucleation, Science, 339, 943–946, 2013. a, b
Kupiainen, O., Ortega, I. K., Kurtén, T., and Vehkamäki, H.: Amine substitution into sulfuric acid – ammonia clusters, Atmos. Chem. Phys., 12, 3591–3599, https://doi.org/10.5194/acp-12-3591-2012, 2012. a
Kürten, A., Jokinen, T., Simon, M., et al.: Neutral molecular cluster formation of sulfuric acid–dimethylamine observed in real time under atmospheric conditions, P. Natl. Acad. Sci. USA, 111, 15019–15024, 2014. a
Kürten, A., Li, C., Bianchi, F., Curtius, J., Dias, A., Donahue, N. M., Duplissy, J., Flagan, R. C., Hakala, J., Jokinen, T., Kirkby, J., Kulmala, M., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Onnela, A., Rissanen, M. P., Simon, M., Sipilä, M., Stozhkov, Y., Tröstl, J., Ye, P., and McMurry, P. H.: New particle formation in the sulfuric acid–dimethylamine–water system: reevaluation of CLOUD chamber measurements and comparison to an aerosol nucleation and growth model, Atmos. Chem. Phys., 18, 845–863, https://doi.org/10.5194/acp-18-845-2018, 2018. a
Kurtén, T., Torpo, L., Sundberg, M. R., Kerminen, V.-M., Vehkamäki, H., and Kulmala, M.: Estimating the NH3 : H2SO4 ratio of nucleating clusters in atmospheric conditions using quantum chemical methods, Atmos. Chem. Phys., 7, 2765–2773, https://doi.org/10.5194/acp-7-2765-2007, 2007. a
Kurtén, T., Loukonen, V., Vehkamäki, H., and Kulmala, M.: Amines are likely to enhance neutral and ion-induced sulfuric acid-water nucleation in the atmosphere more effectively than ammonia, Atmos. Chem. Phys., 8, 4095–4103, https://doi.org/10.5194/acp-8-4095-2008, 2008. a
Larson, L. J., Largent, A., and Tao, M.: Structure of the Sulfuric Acid – Ammonia System and the Effect of Water Molecules in the Gas Phase, J. Phys. Chem. A., 103, 6786–6792, 1999. a
Lawler, M. J., Winkler, P. M., Kim, J., Ahlm, L., Tröstl, J., Praplan, A. P., Schobesberger, S., Kürten, A., Kirkby, J., Bianchi, F., Duplissy, J., Hansel, A., Jokinen, T., Keskinen, H., Lehtipalo, K., Leiminger, M., Petäjä, T., Rissanen, M., Rondo, L., Simon, M., Sipilä, M., Williamson, C., Wimmer, D., Riipinen, I., Virtanen, A., and Smith, J. N.: Unexpectedly acidic nanoparticles formed in dimethylamine–ammonia–sulfuric-acid nucleation experiments at CLOUD, Atmos. Chem. Phys., 16, 13601–13618, https://doi.org/10.5194/acp-16-13601-2016, 2016. a, b
Loeb, N. G. and Kato, S.: Top-of-atmosphere direct radiative effect of aerosols over the tropical oceans from the Clouds and the Earth's Radiant Energy System (CERES) satellite instrument, J. Climate, 15, 1474–1484, 2002. a
Loukonen, V., Kurtén, T., Ortega, I. K., Vehkamäki, H., Pádua, A. A. H., Sellegri, K., and Kulmala, M.: Enhancing effect of dimethylamine in sulfuric acid nucleation in the presence of water – a computational study, Atmos. Chem. Phys., 10, 4961–4974, https://doi.org/10.5194/acp-10-4961-2010, 2010. a, b
Ma, Y., Chen, J., Jiang, S., Liu, Y., Huang, T., Miao, S., Wang, C., and Huang, W.: Characterization of the nucleation precursor (H2SO4-(CH3)2NH) complex: Intra-cluster interactions and atmospheric relevance, RSC Adv., 6, 5824–5836, 2016. a
McMurry, P. H.: The history of condensation nucleus counters, Aerosol Sci. Tech., 33, 297–322, 2000. a
Myllys, N., Chee, S., Olenius, T., Lawler, M., and Smith, J.: Molecular-level understanding of synergistic effects in sulfuric acid–amine–ammonia mixed clusters, J. Phys. Chem. A, 123, 2420–2425, 2019. a
Nadykto, A. B. and Yu, F.: Strong Hydrogen Bonding between Atmospheric Nucleation Precursors and Common Organics, Chem. Phys. Lett., 435, 14–18, 2007. a
Nadykto, A. B., Herb, J., Yu, F., and Xu, Y.: Enhancement in the production of nucleating clusters due to dimethylamine and large uncertainties in the thermochemistry of amine-enhanced nucleation, Chem. Phys. Lett., 609, 42–49, 2014. a
Najibi, A. and Goerigk, L.: The Nonlocal Kernel in van der Waals Density Functionals as an Additive Correction: An Extensive Analysis with Special Emphasis on the B97M-V and ωB97M-V Approaches, J. Chem. Theory Comput., 14, 5725–5738, https://doi.org/10.1021/acs.jctc.8b00842, 2018. a
Neese, F.: Software update: The ORCA program system – Version 5.0, WIREs Comput. Mol. Sci., 12, e1606, https://doi.org/10.1002/wcms.1327, 2022. a
Olenius, T., Kupiainen-Määttä, O., Ortega, I., Kurtén, T., and Vehkamäki, H.: Free energy barrier in the growth of sulfuric acid–ammonia and sulfuric acid–dimethylamine clusters, J. Chem. Phys., 139, 084312, https://doi.org/10.1063/1.4819024, 2013a. a
Olenius, T., Kupiainen-Määttä, O., Ortega, I. K., Kurtén, T., and Vehkamäki, H.: Free energy barrier in the growth of sulfuric acid-ammonia and sulfuric acid-dimethylamine clusters, J. Chem. Phys., 139, 084312, https://doi.org/10.1063/1.4819024, 2013b. a
Ortega, I. K., Kupiainen, O., Kurtén, T., Olenius, T., Wilkman, O., McGrath, M. J., Loukonen, V., and Vehkamäki, H.: From quantum chemical formation free energies to evaporation rates, Atmos. Chem. Phys., 12, 225–235, https://doi.org/10.5194/acp-12-225-2012, 2012. a
Passananti, M., Zapadinsky, E., Zanca, T., Kangasluoma, J., Myllys, N., Rissanen, M. P., Kurtén, T., Ehn, M., Attoui, M., and Vehkamäki, H.: How well can we predict cluster fragmentation inside a mass spectrometer?, Chem. Commun., 55, 5946–5949, 2019. a
Pelucchi, C., Negri, E., Gallus, S., Boffetta, P., Tramacere, I., and La Vecchia, C.: Long-term Particulate Matter Exposure and Mortality: A Review of European Epidemiological Studies, BMC Public Health, 9, 453, https://doi.org/10.1186/1471-2458-9-453, 2009. a
Pracht, P. and Grimme, S.: Calculation of absolute molecular entropies and heat capacities made simple, Chem. Sci., 12, 6551–6568, 2021. a
Pracht, P., Bauer, C. A., and Grimme, S.: Automated and efficient quantum chemical determination and energetic ranking of molecular protonation sites, J. Comput. Chem., 38, 2618–2631, 2017. a
Pracht, P., Bohle, F., and Grimme, S.: Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 22, 7169–7192, 2020. a
Rosenfeld, D., Andreae, M. O., Asmi, A., Chin, M., de Leeuw, G., Donovan, D. P., Kahn, R., Kinne, S., Kivekäs, N., Kulmala, M., Lau, W., Schmidt, K. S., Suni, T., Wagner, T., Wild, M., and Quaas, J.: Global observations of aerosol-cloud-precipitation-climate interactions, Rev. Geophys., 52, 750–808, 2014. a
Sindel, J. P., Gobrecht, D., Helling, C., and Decin, L.: Revisiting Fundamental Properties of TiO2 Nanoclusters as Condensation Seeds in Astrophysical Environments, Astron. Astrophys., 668, A35, https://doi.org/10.1051/0004-6361/202243306, 2022. a
Sipilä, M., Berndt, T., Petäjä, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin, R. L. III, Hyvärinen, A.-P., Lihavainen, H., and Kulmala, M: The role of sulfuric acid in atmospheric nucleation, Science, 327, 1243–1246, 2010. a, b
Spicher, S., Plett, C., Pracht, P., Hansen, A., and Grimme, S.: Automated molecular cluster growing for explicit solvation by efficient force field and tight binding methods, J. Chem. Theory Comput., 18, 3174–3189, 2022. a
Spracklen, D. V., Carslaw, K. S., Kulmala, M., Kerminen, V.-M., Mann, G. W., and Sihto, S.-L.: The contribution of boundary layer nucleation events to total particle concentrations on regional and global scales, Atmos. Chem. Phys., 6, 5631–5648, https://doi.org/10.5194/acp-6-5631-2006, 2006. a
Temelso, B., Morrison, E. F., Speer, D. L., Cao, B. C., Appiah-Padi, N., Kim, G., and Shields, G. C.: Effect of mixing ammonia and alkylamines on sulfate aerosol formation, J. Phys. Chem. A, 122, 1612–1622, 2018. a
Vanhanen, J., Mikkilä, J., Lehtipalo, K., Sipilä, M., Manninen, H., Siivola, E., Petäjä, T., and Kulmala, M.: Particle size magnifier for nano-CN detection, Aerosol Sci. Tech., 45, 533–542, 2011. a
Weigend, F. and Ahlrichs, R.: Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 7, 3297–3305, https://doi.org/10.1039/B508541A, 2005. a
Wilemski, G. and Wyslouzil, B. E.: Binary nucleation kinetics. I. Self-consistent size distribution, J. Chem. Phys., 103, 1127–1136, 1995. a
Wu, H., Engsvang, M., Knattrup, Y., Kubecka, J., and Elm, J.: Improved Configurational Sampling Protocol for Large Atmospheric Molecular Clusters, ACS Omega, 8, 45065–45077, 2023. a, b, c, d, e
Wu, H., Knattrup, Y., Jensen, A. B., and Elm, J.: Cluster-to-particle transition in atmospheric nanoclusters, Aerosol Research, 2, 303–314, https://doi.org/10.5194/ar-2-303-2024, 2024. a, b, c, d, e, f, g, h, i, j, k
Yu, H., McGraw, R., and Lee, S.-H.: Effects of amines on formation of sub-3 nm particles and their subsequent growth, Geophys. Res. Lett., 39, L02807, https://doi.org/10.1029/2011GL050099, 2012. a
Zapadinsky, E., Passananti, M., Myllys, N., Kurtén, T., and Vehkamäki, H.: Modeling on fragmentation of clusters inside a mass spectrometer, J. Phys. Chem. A, 123, 611–624, 2018. a
Zhang, J. and Dolg, M.: ABCluster: the artificial bee colony algorithm for cluster global optimization, Phys. Chem. Chem. Phys., 17, 24173–24181, https://doi.org/10.1039/C5CP04060D, 2015. a
Zhang, J. and Dolg, M.: Global optimization of clusters of rigid molecules using the artificial bee colony algorithm, Phys. Chem. Chem. Phys., 18, 3003–3010, https://doi.org/10.1039/C5CP06313B, 2016. a
Zheng, J., Xu, X., and Truhlar, D. G.: Minimally augmented Karlsruhe basis sets, Theor. Chem. Acc., 128, 295–305, https://doi.org/10.1007/s00214-010-0846-z, 2011. a, b