the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 28 Feb 2025
| 28 Feb 2025
Uptake of organic vapours and nitric acid on atmospheric freshly nucleated particles
Yosef Knattrup
Sulfuric acid, ammonia, and amines are believed to be key contributors to the initial steps in new particle formation in the atmosphere. However, other compounds such as organic compounds or nitric acid are believed to be important for further growth at larger sizes. In this study, we investigate the potential uptake of first-generation oxidation products from α-pinene (pinic and pinonic acid) and isoprene (trans-β-IEPOX, β4-ISPOOH, and β1-ISOPOOH), a potential highly oxidised molecule (HOM), formic acid, and nitric acid. The uptake is probed onto (SA)10(base)10 freshly nucleated particles (FNPs), where SA denotes sulfuric acid, and the bases are ammonia (AM), methylamine (MA), dimethylamine (DMA), or trimethylamine (TMA). The addition free energies were calculated at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory. We find favourable addition free energies of −8 to −10 kcal mol−1 for the HOM, pinic acid, and pinonic acid on the less sterically hindered (SA)10(AM)10 and (SA)10(MA)10 FNPs. This suggests that isoprene oxidation products do not contribute to the early growth of FNPs, but the α-pinene products do, in accordance with their expected volatilities.
Calculating the second addition of a pinic acid molecule or pinonic acid molecule on the (SA)10(AM)10 FNPs, we find that pinic acid maintains its large addition free energy decrease due to its two carboxylic acid groups interacting with the other monomer, as well as the FNP. The pinonic-acid addition free energy drops to −3.9 kcal mol−1 due to the weak interactions between the FNP and its carbonyl group and the lack of monomer–monomer interactions. Calculating the addition free energy under realistic atmospheric conditions, we find that the FNPs studied are too small (1.4 nm) to support the growth of the studied uptake monomers. We find that the accretion product pinyl diaterpenylic ester (PDPE; C17H26O8) yields an addition free energy value of −17.1 kcal mol−1. This suggests that PDPE can overcome the strong Kelvin effect of a 1.4 nm FNP and lead to spontaneous uptake under ambient conditions.
- Article
(7782 KB) - Full-text XML
- BibTeX
- EndNote




Clouds play a significant role in shaping the global climate, and their formation begins when water condenses onto cloud condensation nuclei (CCN) (Boucher and Lohmann, 1995). Aerosols can act as CCN, delivering the required surface area for water to condense on, when they reach sizes of roughly 50–100 nm in diameter (Boucher and Lohmann, 1995). The cloud–aerosol interaction is, according to the Intergovernmental Panel on Climate Change (IPCC) Fifth Assessment Report, the largest cause of uncertainty in modern radiative forcing modelling (Canadell et al., 2021). Aerosols form through two main pathways, namely either as primary particles, directly emitted into the atmosphere, or as secondary particles, which form through the clustering of gas vapours (Kulmala et al., 2013). Around 50 % of CCN are believed to originate from the secondary process (Boucher and Lohmann, 1995; Merikanto et al., 2009), denoted new particle formation (NPF). As an example, Zhao et al. (2024) simulated the spatial distribution of CCN at the surface level and found the fraction of CCN from NPF to be around 30 %–40 % over mainland Europe and up to ≈ 60 % over the eastern United States.
The substantial uncertainty in the climate models is linked with the unidentified mechanisms for the NPF pathways and the early growth behaviour of freshly nucleated particles (FNPs) (Canadell et al., 2021). For instance, Tröstl et al. (2016) found a factor of 2 difference in predicted CCN number concentration when altering the 1.7–3.0 nm particle growth mechanisms in their global model. It is therefore important to elucidate the initial growth mechanisms. Wang et al. (2020) found, experimentally, that vapours of ammonia (AM) and nitric acid (NA) could condense onto freshly nucleated particles (FNPs) at 278.5 K, but temperatures below 258.15 K were required for the nucleation of NA and AM. They extended this study (Wang et al., 2022) to include sulfuric acid (SA) and found that NA, SA, and AM could form particles under conditions similar to the upper free troposphere. Stolzenburg et al. (2018) studied the influence of organics in early particle growth in the CLOUD chamber and found rapid growth from organics in temperature ranges of 248.15–298.15 K.
Experimentally, the main focus has been on measuring particles larger than 2 nm in diameter, as particles below this size are challenging to measure accurately. Sub-2 nm charged-cluster compositions have been measured by mass spectrometry techniques (Jokinen et al., 2012); however, it is uncertain if the measured clusters underwent fragmentation inside the instrument or if the charging of neutral clusters changes the composition during the measurement (Zapadinsky et al., 2019; Passananti et al., 2019). This positions quantum chemical methods as a key tool for exploring the formation of small clusters and their early growth processes.
The initial clustering process is believed to primarily be driven by sulfuric acid (SA) stabilised by ammonia (AM) (Kulmala et al., 2013; Kirkby et al., 2011; Schobesberger et al., 2013; Weber et al., 1996; Elm, 2021a; Dunne et al., 2016; Kubečka et al., 2023b) or strong amines such as methylamine (MA) (Kurtén et al., 2008; Nadykto et al., 2011, 2015, 2014; Jen et al., 2014; Glasoe et al., 2015; Kubečka et al., 2023b), dimethylamine (DMA) (Kurtén et al., 2008; Loukonen et al., 2010; Nadykto et al., 2011, 2015, 2014; Jen et al., 2014; Glasoe et al., 2015; Almeida et al., 2013; Elm, 2021a; Kubečka et al., 2023b), and trimethylamine (TMA) (Elm, 2021a; Kurtén et al., 2008; Nadykto et al., 2011; Jen et al., 2014; Nadykto et al., 2015; Glasoe et al., 2015; Kubečka et al., 2023b). Nitric acid (NA) (Wang et al., 2020; Liu et al., 2021, 2018; Kumar et al., 2018; Ling et al., 2017; Wang et al., 2022; Nguyen et al., 1997; Longsworth et al., 2023; Knattrup et al., 2023; Knattrup and Elm, 2022; Bready et al., 2022; Qiao et al., 2024) and formic acid (FA) (Bready et al., 2022; Knattrup et al., 2023; Ayoubi et al., 2023; Zhang et al., 2022, 2018; Harold et al., 2022; Nadykto and Yu, 2007) have also been shown to be involved in the initial clustering. Organics have not been definitively proven to take part in the initial clustering process, but they are known to be important for the growth (Kulmala et al., 2013; Elm et al., 2020, 2023; Engsvang et al., 2023b).
While much of the focus has been on the initial clustering process (up to eight monomers) (Elm et al., 2020, 2023; Engsvang et al., 2023b), Engsvang et al. (2023a), Engsvang and Elm (2022) and Wu et al. (2023, 2024) have pushed the studies up to large particles of up to 30–60 monomers, reaching geometric diameters of up to 2 nm, where the clusters start exhibiting more particle-like properties. Wu et al. (2024) defined the cluster-to-particle transition point as the point where the free energy per monomer starts levelling off, thereby resembling “bulk” thermodynamics and where “solvated” monomers appear in the cluster structures. They found this point to be around 10 acid–base pairs for the SA–AM/MA/DMA/TMA clusters and defined the clusters at and beyond this point as FNPs. Previously, DePalma et al. (2015) studied the uptake of pinic and pinonaldehyde on a (SA)4(AM)4 cluster at the AM1 level of theory. However, the early growth of realistic sizes (∼ 2 nm) of FNPs has yet to be studied.
In this paper, we study the uptake of NA and common organics (denoted X) on the (SA)10(AM/MA/DMA/TMA)10 FNPs using quantum chemical methods for the following reaction scheme:
We chose these sizes as the clusters have all reached the cluster-to-particle transition point. We investigate the first-generation oxidation products of α-pinene, isoprene (Nozière et al., 2015), and a potential highly oxidised molecule (HOM) of α-pinene (Kurtén et al., 2016) (as suggested by COSMO-RS calculations). The studied systems are displayed in Fig. 1.
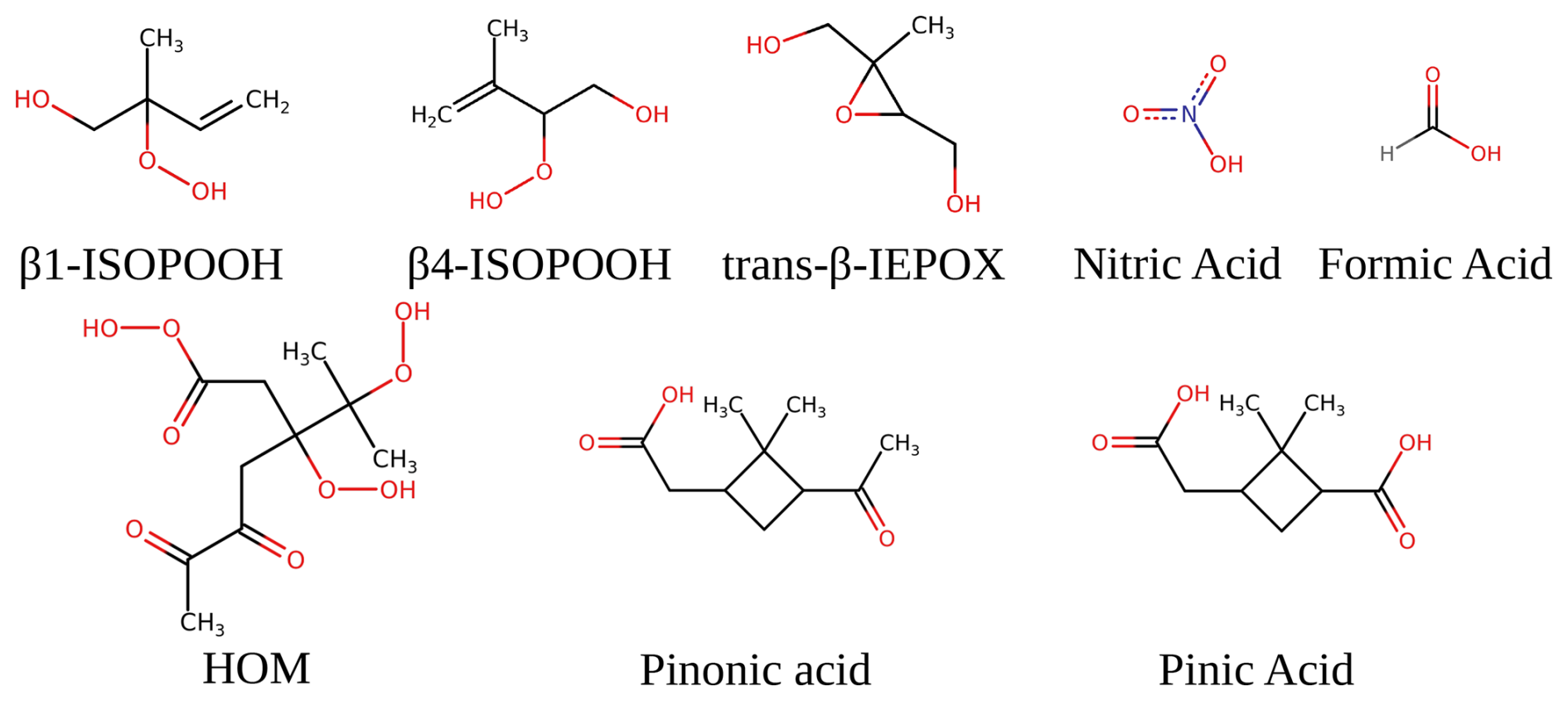
Figure 1The molecular structure of the studied monomers. Pinonic and pinic acid are first-generation oxidation products from α-pinene. Moreover, trans-β-IEPOX, β4-ISOPOOH, and β1-ISOPOOH are first-generation oxidation products from isoprene.
2.1 Computational details
The B97-3c (Brandenburg et al., 2018) and ωB97X-D3BJ//6-311++G(3df,3pd) (Najibi and Goerigk, 2018; Ditchfield et al., 1971) calculations were performed in ORCA 5.0.4 (Neese, 2012; Neese et al., 2020; Neese, 2022). The xTB 6.4.0 (Bannwarth et al., 2021) program was used for the GFN1-xTBre-par calculations. GFN1-xTBre-par is a parameterisation of GFN1-xTB (Grimme et al., 2017), using the methodology suggested by Knattrup et al. (2024). This specific parameterisation was done by Wu et al. (2024), where a linear combination of the binding energy and gradient errors were minimised on a set of B97-3c FNPs. Configurational sampling was performed using ABCluster 3.2 (Zhang and Dolg, 2015, 2016; Zhang, 2022) and CREST 2.12 (Grimme, 2019; Pracht et al., 2020) in noncovalent interaction mode. The entire computational workflow and subsequent data handling were performed using the JKCS 2.1 suite of programs (Kubečka et al., 2023a). All the data are freely available in the atmospheric cluster database (Kubečka et al., 2023a; Elm, 2019). See the “Data availability” section for more information.
2.2 Thermochemistry
We define the binding free energy ΔGbind as the free energy change between the combined cluster (Gcluster) and the free energy (Gmonomer) from the n monomers as follows:
To get more accurate free energies, we performed a higher-level single-point correction on top of the density functional theory (DFT) geometries. This is denoted with the sp//geo notation, where “sp” is the single-point energy method used to calculate the electronic energy (Esp), and “geo” is the method used to calculate the thermal correction to the free energy () from the optimised structure and vibrational frequencies. The thermal correction term includes everything, except the electronic energy,
The addition free energy is the binding free energy change when a monomer is added to the cluster as follows:
We employ the quasi-harmonic approximation (Grimme, 2012) (as standard in ORCA), where vibrational frequencies below 100 cm−1 are treated as free-rotor contributions to the entropy.
where kB is the Boltzmann constant, T the temperature, I′ the effective moment of inertia, R the gas constant, and h the Planck constant.
The output of quantum chemical programs is, for the most part, under standard conditions (p = 1 atm; T = 298.15 K). Changing the temperature can be done by re-evaluating the standard statistical mechanics expressions at the wanted temperature. If the binding free energy is needed under other conditions, Halonen (2022) derived the following equation for the binding free energy at the given monomer concentrations:
where pi is the partial pressure (concentration) of monomer i, n is the number of monomers, and pi is the reference pressure at which the reference binding free energy was calculated. This equation correctly satisfies self-consistency for multi-component clusters (the monomers will have a free energy of zero) and the law of mass action. Combining Eq. (6) with Eq. (4) yields the addition free energy under the given conditions, namely
where n is the total number of monomers in the largest cluster, is the reference addition free energy, the sum runs over all monomers in the clusters before the addition, and padd is the pressure (concentration) of the added monomer.
2.3 Configurational sampling workflow
We employed the configurational workflow for FNPs as suggested by Wu et al. (2023, 2024).
The workflow entails 10 parallel runs of ABCluster (Zhang and Dolg, 2015, 2016), with SN = 1280, gen = 320, and sc = 4. We used matching ionic monomers, leading to an overall electronic neutral cluster. We choose to have ionic inorganic monomers and neutral organic monomers. We chose this because SA is a stronger acid compared to the monomers. All the structures from ABCluster are subsequently optimised with GFN1-xTBre-par and have a single-point energy calculated at the B97-3c level of theory. The 1000 structures that are lowest in electronic energy are then partially optimised with B97-3c for 40 iterations. The 100 structures that are lowest in electronic energy are then fully optimised, and finally, a vibrational frequency calculation is performed.
The lowest free energy is used as the input structure for CREST, using GFN1-xTBre-par, as suggested by Knattrup et al. (2024), and the 100 structures that are lowest in electronic energy are then fully optimised, followed by a vibrational frequency calculation.
The larger clusters suffered from memory issues and SCF convergence problems, causing a low number of fully optimised structures using our automated workflow. We, therefore, restarted all 200 cluster calculations with increased memory and VeryTightSCF and redid the CREST workflow, resulting in some cluster systems having over 200 configurations. The organic monomers were sampled using CREST. The ABCluster input was generated from the lowest energy conformer, using topgen (Zhang, 2022; Zhang and Dolg, 2015, 2016) with charges from an MP2/6-31++G calculation in Gaussian 16 (Gaussian et al., 2016).
2.4 Single-point refinement
Engsvang et al. (2023a) found that the B97-3c single-point energies gave quite erroneous addition free energies for FNPs compared to the DLPNO-CCSD(T0)/aug-cc-pVTZ reference calculations. They found that ωB97X-D3BJ/6-311++G(3df,3pd) had excellent error cancellation for the addition free energies yielding errors below 1 kcal mol−1. Hasan et al. (2024) extended the benchmark by including single-point calculations with ωB97X-D3BJ and the augmented def2 basis sets. However, none managed to beat the combination found by Engsvang et al. (2023a). We, therefore, performed ωB97X-D3BJ/6-311++G(3df,3pd) single-point energy calculations on the 10 lowest free energy configurations. The SCF calculations had trouble converging due to linear dependencies in the basis; therefore, we had to increase the scfthres parameter to . This change slightly increases the energy (max of 0.6 kcal mol−1 for the 10 acid 10 base systems); however, it is systematic for the given system size and should partly cancel out for the binding addition free energies.
It should be noted that the ωB97X-D3BJ/6-311++G(3df,3pd) single-point energies slightly overbind (roughly 4 kcal mol−1) compared to the DLPNO-CCSD(T0)/aug-cc-pVTZ reference level by Engsvang et al. (2023a). However, all the energies are roughly shifted in the same manner, leading to less erroneous addition free energies.
3.1 Addition free energies
To probe the potential for the (SA)10(AM/MA/DMA/TMA)10 FNPs to uptake monomers (pinic and pinonic acid, trans-β-IEPOX, β4-ISPOOH, and β1-ISOPOOH), HOM, FA, and NA, we calculated the addition free energies as defined in Eq. (4).
The reference FNPs, without the uptake monomers, are taken from Wu et al. (2024). They found that, for realistic atmospheric conditions ([SA] = 106 molec. cm−3; AM = (10 ppt, 10 ppb); MA/DMA/TMA = (1, 10 ppt)), the formation of the SA–AM and SA–MA FNPs go through nucleation barriers but are stable after the barrier. Furthermore, the formation of the SA–DMA FNP is found to be entirely barrierless, but the formation of the SA–TMA FNP is unfavourable (due to steric hindrance) as the binding free energy is positive for the studied conditions.
Figure 2 presents the calculated addition free energies at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory. It should be noted that the most negative binding free energy of the FNPs follows the order of MA > DMA > AM > TMA. This trend does not align with the gas-phase acidity or basicity of the components. Instead, steric factors become increasingly important for larger sizes, thus changing the order. In the following sections, we will discuss the trends for each FNP type.
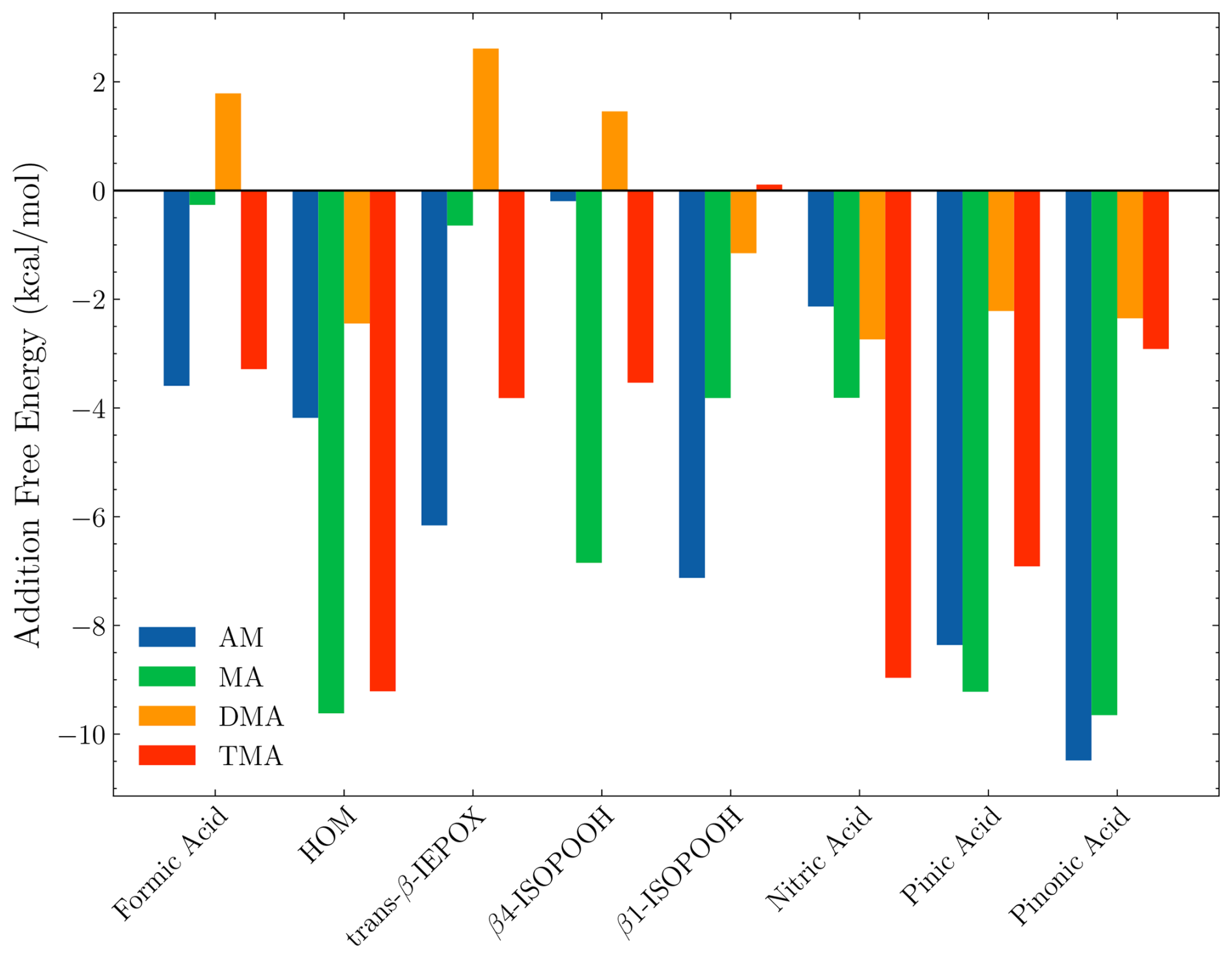
Figure 2The addition free energy as defined in Eq. (4). The free energy is calculated using the lowest Gibbs free configuration (standard conditions) at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory. The x axis shows the monomer added to the (SA)10(base)10 FNP, where the label defines the base.
3.1.1 SA–DMA
From Fig. 2, it is seen that the (SA)10(DMA)10 FNPs are the least favourable for the uptake of vapours, with positive addition free energies for FA, IEPOX, and β4-ISOPOOH and higher than −3 kcal mol−1 addition free energies for the remaining monomers. This is due to the several effects. First, SA–DMA FNPs are already substantially more stable than the other FNPs due to the high basicity of DMA compared to the other bases (Wu et al., 2024). Hence, the reference system in Eq. (4) is already low in free energy, leading to higher addition free energies. Second, the two bulky methyl groups in the DMA molecules will lead to some steric hindrance when adding the organics to the FNP. Third, adding the new monomer disrupts the already favourable hydrogen bond network of the SA–DMA FNPs, either by destabilising the system or by only making it slightly more favourable. This concept is illustrated in Fig. 3 for the (SA)10(DMA)10(trans-β-IEPOX)1 structure. The addition of the IEPOX molecule forms an epoxide–DMA bond (light-blue circle), which forces the methyl group on the DMA molecule slightly inside the cluster, leading to increased steric repulsion. This is in contrast to the preferred orientation of the DMA molecule, where both methyl groups point out of the cluster (dark-blue square).
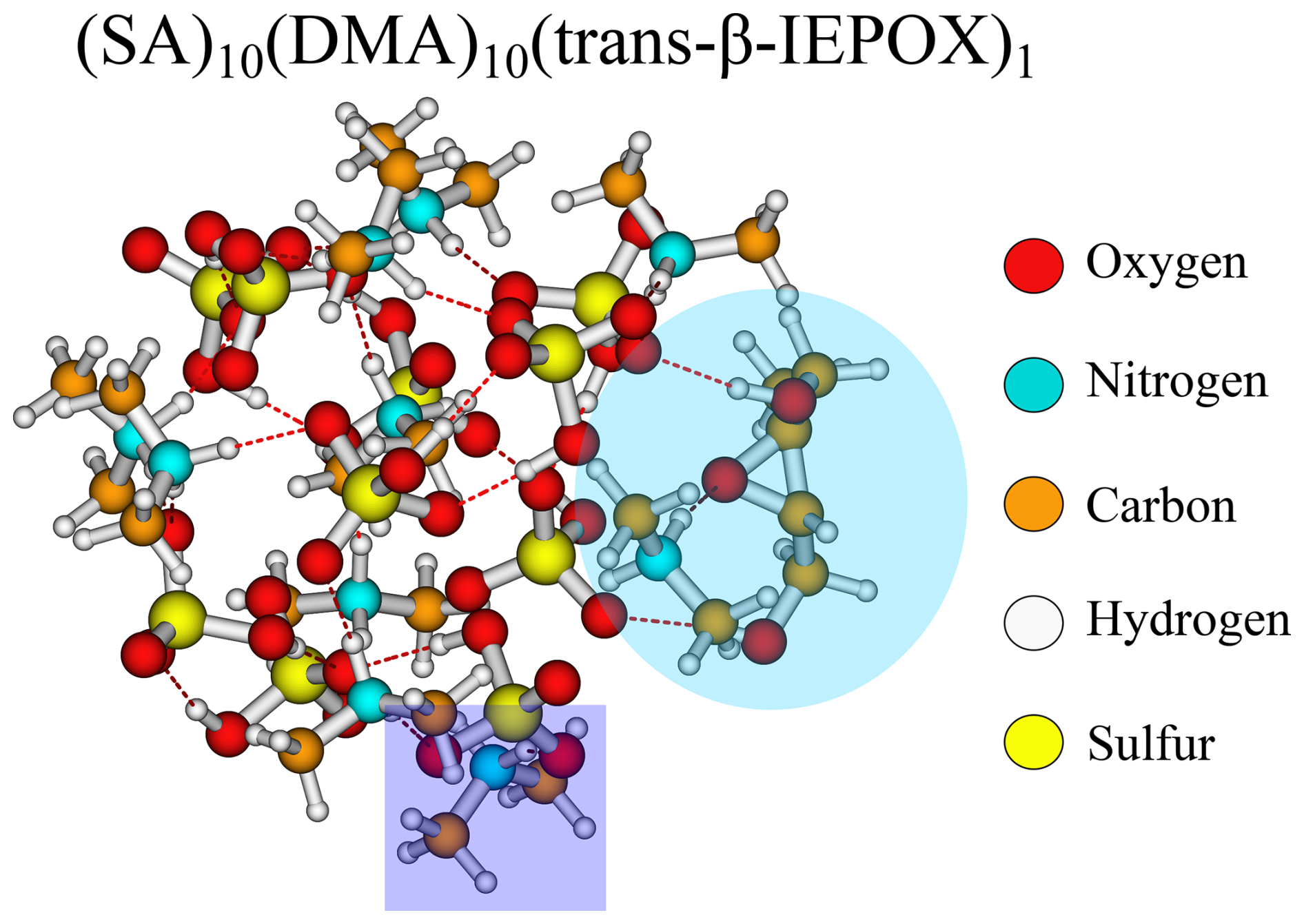
Figure 3The (SA)10(DMA)10(trans-β-IEPOX)1 cluster lowest in free energy (standard conditions) at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
3.1.2 SA–TMA
For the (SA)10(TMA)10 FNPs, all the addition free energies are negative, except for β1-ISOPOOH (+0.1 kcal mol−1). This is mainly caused by the fact that the SA–TMA FNPs by themselves are relatively unstable (Wu et al., 2024). Although TMA is a stronger base in terms of gas-phase basicity (Hunter and Lias, 1998) compared to DMA, its three bulky methyl groups and only a single hydrogen bond donor prevents it from obtaining a favourable hydrogen bond topological network. Hence, introducing an additional monomer extends the hydrogen bond network and lowers the steric hindrance. It is curious that there is such a large difference between the two isomers of ISOPOOH (+0.1 kcal mol−1 vs. −3.5 kcal mol−1) as they are structurally very similar (see Fig. 4). The geometries cannot easily explain the difference as their carbon backbones point outwards from the cluster and do not interact with the “core”. This leaves the main cluster geometry as the driving parameter. Here, the (SA)10(TMA)10(β4-ISOPOOH)1 cluster seems a bit less tightly packed compared to the β1-ISOPOOH cluster, which might equal a more favourable hydrogen bonding topological network.
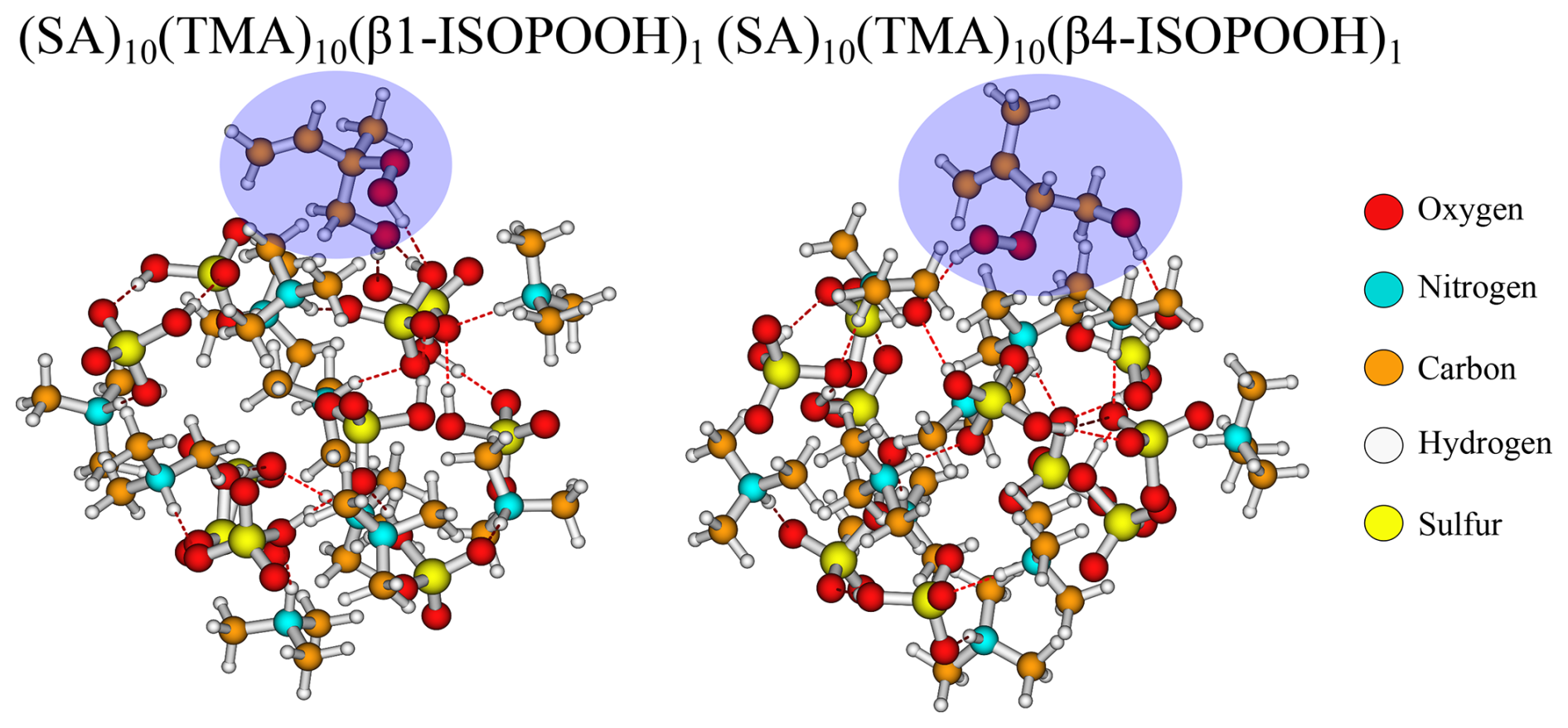
Figure 4The (SA)10(TMA)10(β1-ISOPOOH)1 and (SA)10(TMA)10(β4-ISOPOOH)1 cluster lowest in free energy (standard conditions) at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
Likewise, it is also curious that the bulky HOM has addition free energy in similar magnitudes (−9.2 kcal mol−1) to the much smaller nitric acid (−9.0 kcal mol−1) and pinic acid (−6.9 kcal mol−1). Studying the structure (Fig. 5), the HOM appears to compress the entire cluster, “hovering” slightly above the surface and favouring internal hydrogen bonding as seen on top of the HOM. This means that, while the first addition is very favourable, the second addition of another HOM is less likely as this would potentially compress the cluster even further. However, the internal hydrogen bonds in the HOM could potentially be broken to act as a tether between two adjacent clusters.
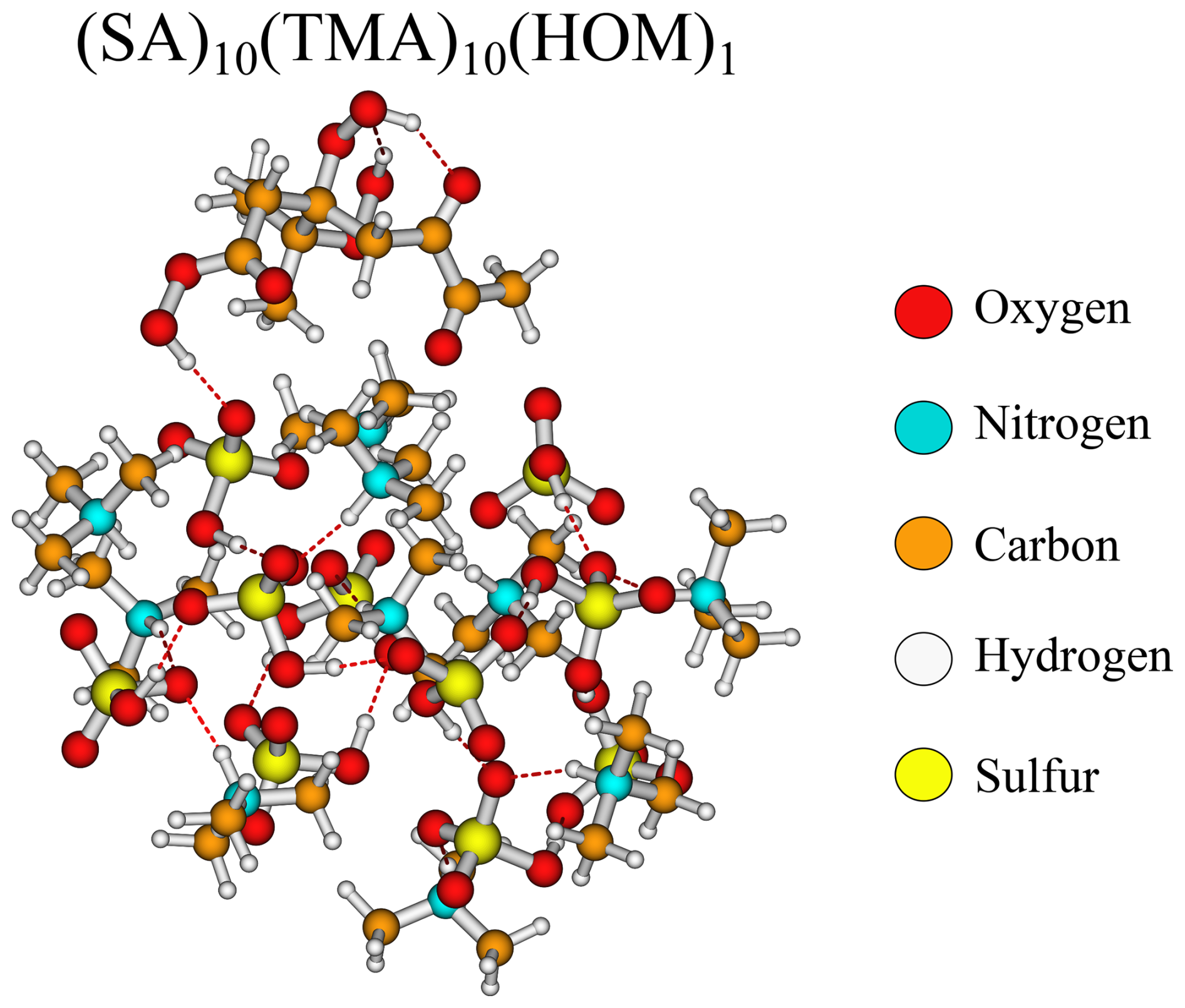
Figure 5The (SA)10(TMA)10(HOM)1 cluster lowest in free energy (standard conditions) at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
3.1.3 SA–MA
The SA–MA FNPs have a notable negative addition free energy for the HOM (−9.6 kcal mol−1), pinic (−9.2 kcal mol−1), and pinonic acid (−9.7 kcal mol−1). It is interesting that they are almost equivalent as pinic acid has two carboxylic acid groups, whereas in pinonic acid one of the carboxylic acid groups is exchanged for a carbonyl group, and we would expect carboxylic acid to bind stronger.
Studying the structures (Fig. 6), we see that they reside at the surface of the cluster, interacting with their carboxylic acid/carbonyl group in almost the same way. This suggests the clusters can rearrange to accommodate both types of interactions, and it does not matter much that one of the carboxylic acid groups has been exchanged with a carbonyl group.
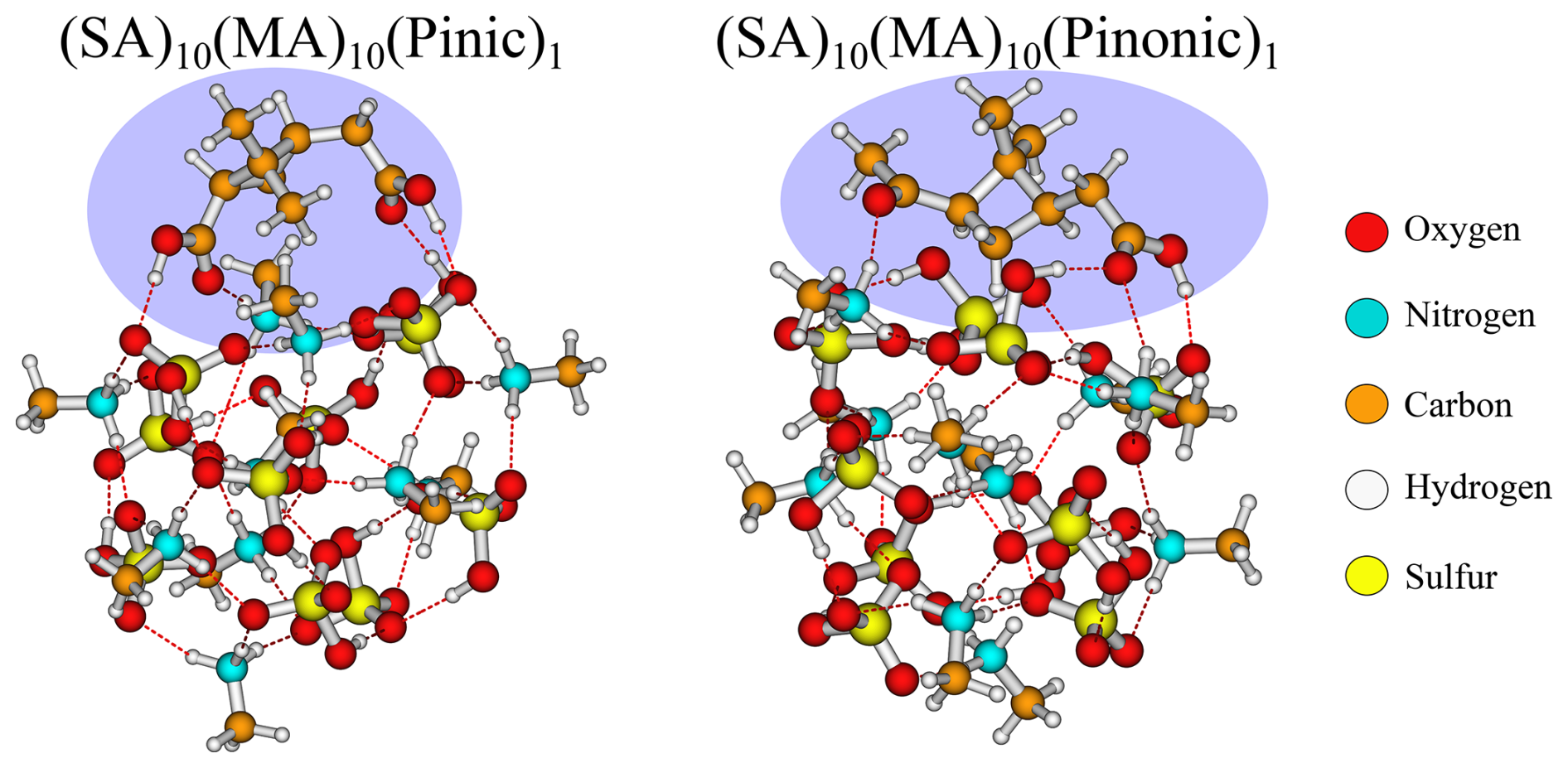
Figure 6The (SA)10(MA)10(pinic)1 and (SA)10(MA)10(pinonic)1 cluster lowest in free energy (standard conditions) at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
Like the SA–TMA cluster, it is quite favourable to add the HOM (−9.6 kcal mol−1), but, unlike the SA–TMA structure, this matches chemical intuition as the HOM almost maximises the interactions with the cluster, as seen in Fig. 7. This also fits with the interaction pattern of pinic and pinonic acid, thus explaining the magnitude of the addition free energy.
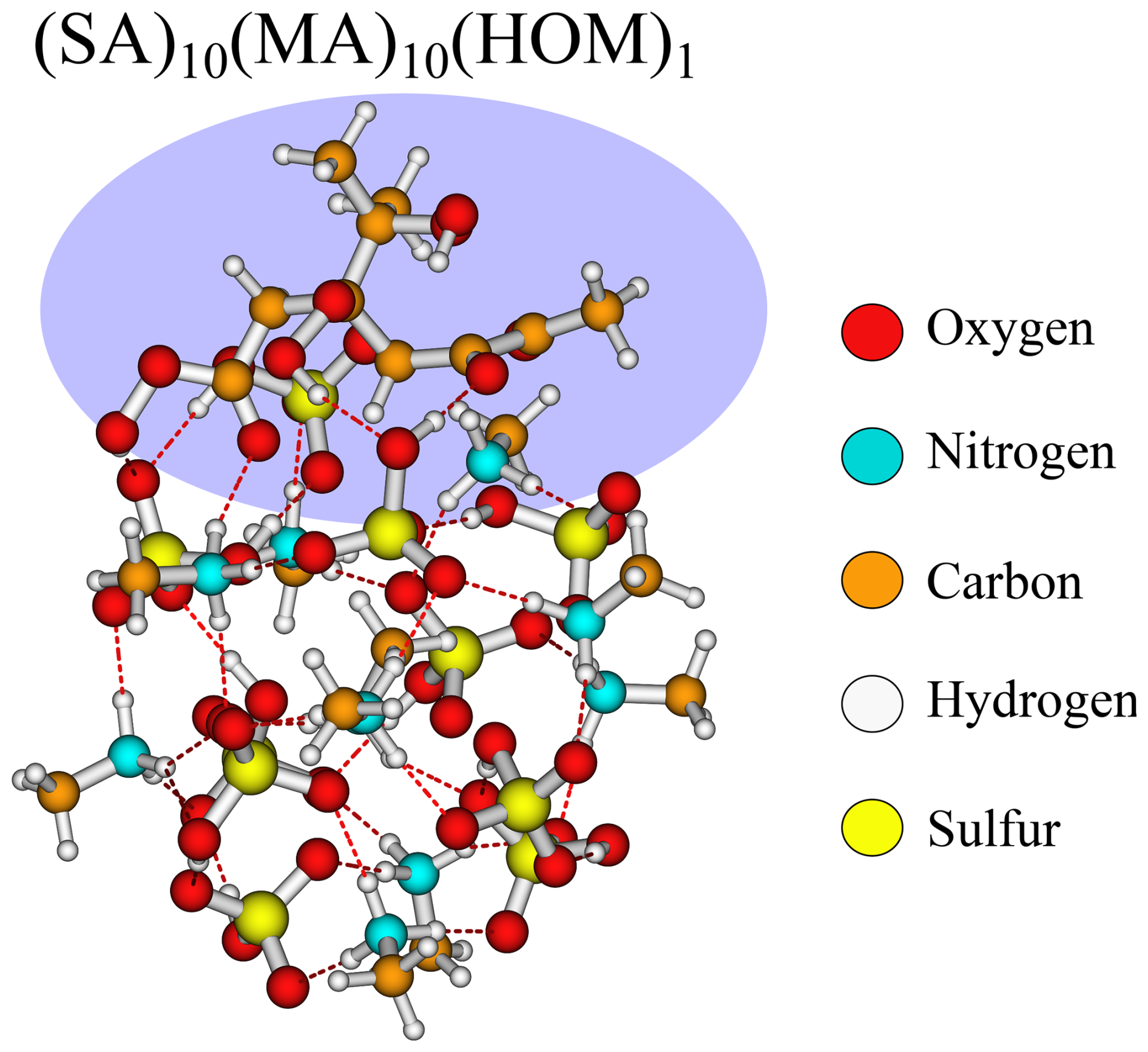
Figure 7The (SA)10(MA)10(HOM)1 cluster lowest in free energy (standard conditions) at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
3.1.4 SA–AM
The SA–AM FNPs have a similar negative addition free energy for pinic (−8.4 kcal mol−1) and pinonic acid (−10.5 kcal mol−1) to the SA–AM FNP. They also have the same structural characteristics. However, there is an inverse trend for the remaining monomers (FA, HOM, trans-β-IEPOX, β4-ISOPOOH, β1-ISOPOOH, and NA), namely if the addition free energy is substantially negative for one type of FNP, then it is small for the other type of FNP for a given monomer. This is especially noticeable for the β4-ISOPOOH monomer, where the addition free energy is almost zero for the SA–AM FNP but ≈ −7 kcal mol−1 for the SA–MA FNP. Structurally, it is unclear why the addition free energy differs so much as both interact with the cluster's outer layer, and neither of the “backbone” carbon chains interacts with the cluster (Fig. 8; ISOPOOH has been marked with a dark-blue box). The only major difference is that, in the SA–AM FNP, the dihedral angle between the two functional groups in ISOPOOH allows it to attach entirely to the surface, while for the SA–MA FNP, the dihedral angle between the functional groups causes it to be slightly more embedded in the cluster.
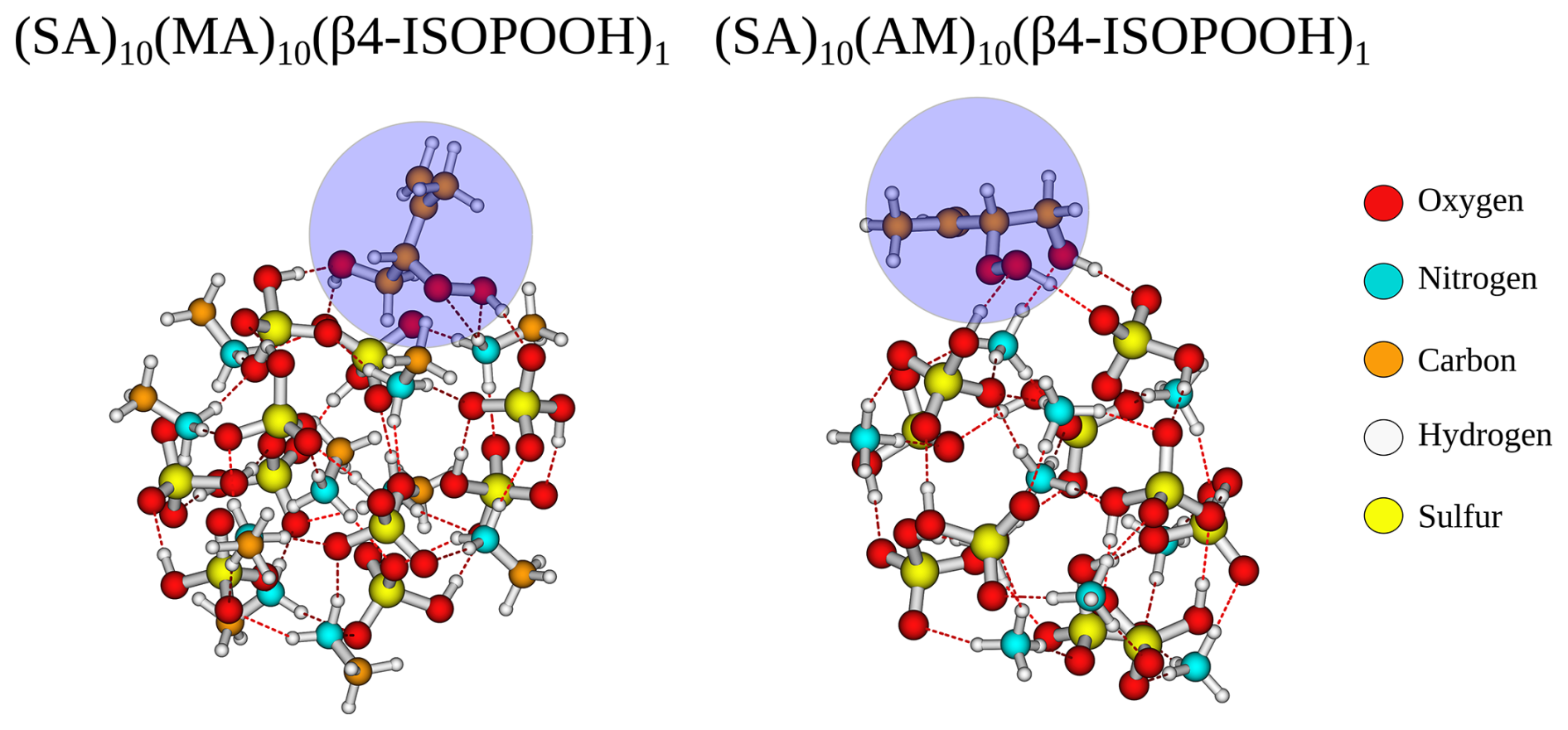
Figure 8The (SA)10(MA)10β4-ISPOOH)1 and (SA)10(AM)10β4-ISPOOH)1 cluster lowest in free energy (standard conditions) at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
3.2 Potential for organic growth
It is clear that FNPs containing 10 acid–base pairs might have the potential to grow via organic uptake, as we observed addition free energies below −8 kcal mol−1 for the HOM, pinic acid, and pinonic acid. It appears that isoprene oxidation products have a lower potential for contributing to early growth compared to the α-pinene products. This is in accordance with the expected vapour pressure of the compounds.
Out of the studied organic compounds, the HOM and pinic and pinonic acid seem to be the most likely candidates for organic growth. To further investigate this, we calculated the addition free energy of a second pinic and pinonic acid molecule. We chose to study the SA–AM FNP, as it showed a large potential for the uptake of these species.
The addition free energy of the second addition is −11.8 and −3.9 kcal mol−1 for pinic and pinonic acid, respectively. The addition-free energy of pinic acid is even lower than the first (−8.4 kcal mol−1). By contrast, the addition free energy of the second pinonic acid increased dramatically from the first addition (−10.5 kcal mol−1). This stark contrast can easily be seen from the geometries in Fig. 9. Pinic acid, with its two carboxylic acid groups, can not only bind with the surface of the FNP but also with the other pinic acid. One could envision that with further additions of pinic acid molecules, they could link together over the entire surface, essentially leading to a core-shell structure. In contrast, pinonic acid only has one carboxylic acid group which attaches to the surface. However, it does not link the two monomers together as the carbonyl groups favours binding with the bases in the FNP. This is in agreement with the earlier findings by Elm et al. (2017) and the cluster-of-functional-group approach by Pedersen et al. (2024), which states that carboxylic acid groups yield the most negative addition free energy for organic-enhanced atmospheric cluster formation. The fact that the organic–organic interaction is affecting the FNP growth is an intriguing result. This could indicate that the uptake of organics on small particles, and by extension the growth thermodynamics, significantly deviates from the usually employed metric of the saturation vapour pressure of the organics. Hence, uptake of organics might be as dependent on the organic–organic interactions as the organic–FNP interactions. This implies that the co-condensation of various vapours on FNPs will depend strongly on the exact functional groups in the molecules and should be further studied in the future.
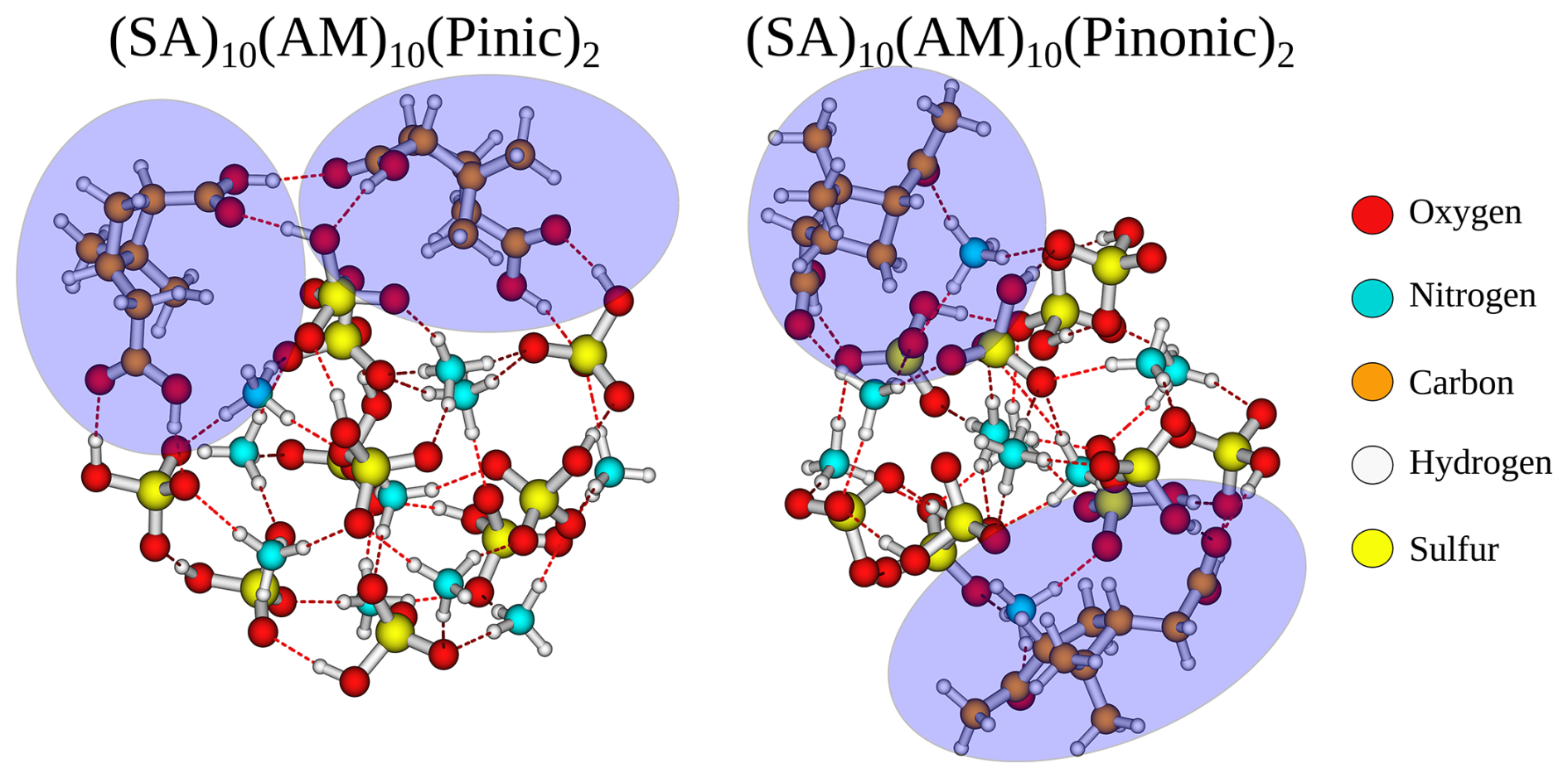
Figure 9The (SA)10(AM)10(pinic)2 and (SA)10(AM)10(pinonic)2 cluster lowest in free energy (standard conditions) at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
3.3 Free energies under actual conditions
The free energies calculated in the previous sections are under standard conditions (p = 1 atm; T = 298.15 K). However, as none of the species involved is present in such high concentrations in the atmosphere, the sign of the standard free energy change alone does not determine whether the addition is spontaneous under atmospheric conditions. To explore this, we scanned the free energy dependence on temperature and monomer concentration for the addition of pinic acid onto the (SA)10(AM/MA)10 FNPs. These systems were chosen because they showed favourable standard free energy changes for the first (and second, in the case of AM) addition. Two concentration regimes for the FNP components were selected based on the “clusteromics” series of papers (Elm, 2021a, b, 2022; Knattrup and Elm, 2022; Ayoubi et al., 2023). For both regimes, SA is set to 106 molec. cm−3, and the concentrations of bases were AM (10 ppt; 10 ppb), MA (1, 100 ppt) and DMA/TMA (1, 10 ppt) for the lower and upper regimes, respectively. Ambient pinic acid concentrations were assumed to range from 0.5 to 20.5 ppt.
Figure 10 shows the maximum temperature at which the addition of pinic acid to either (SA)10(AM)10 or (SA)10(MA)10 becomes spontaneous across these concentration regimes.
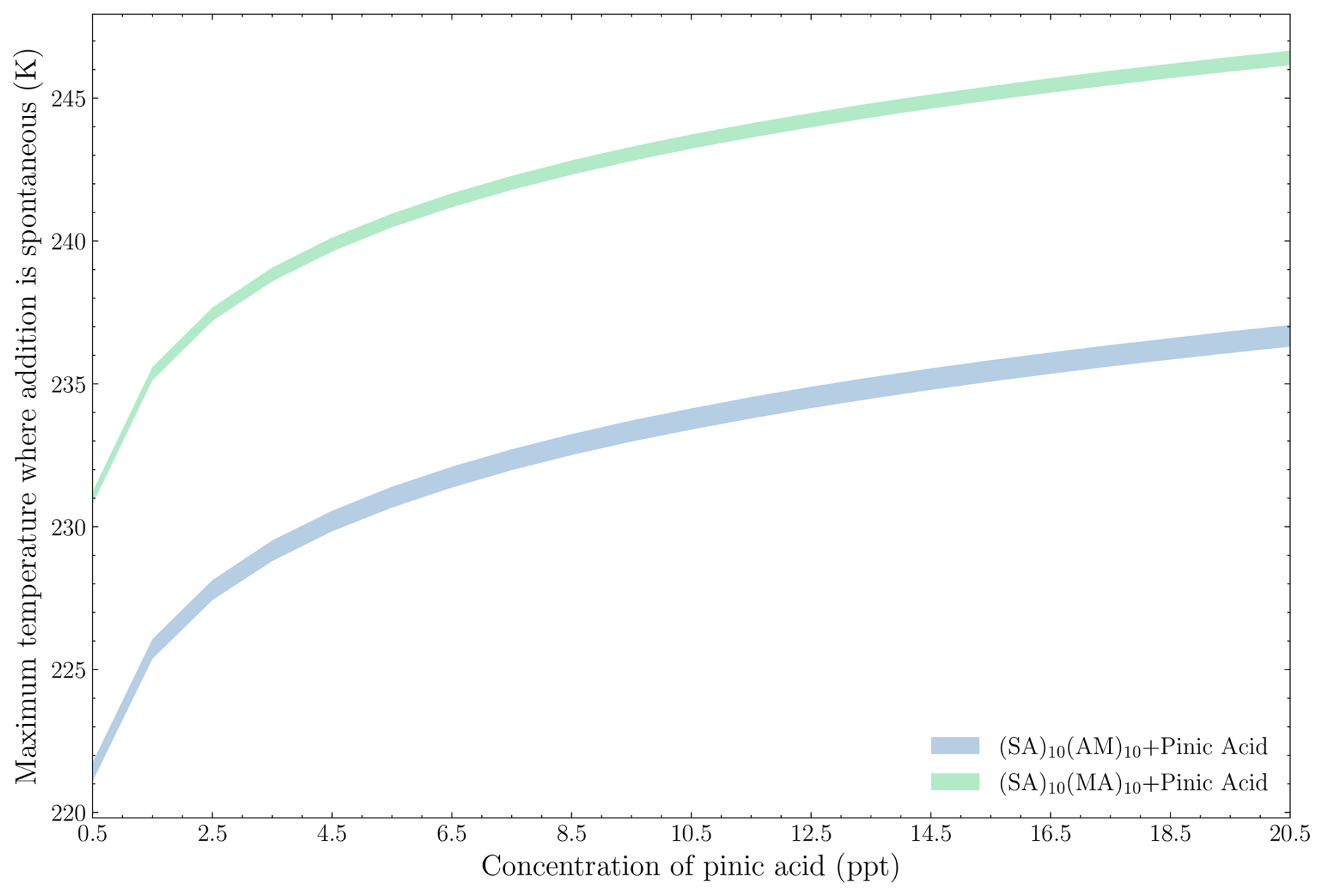
Figure 10The maximum temperature at which the addition of pinic acid onto either (SA)10(AM)10 or (SA)10(MA)10 is spontaneous under the given conditions, ranging from the upper- to the lower-concentration regime for the bases (top of distribution = upper concentration regime; bottom of distribution = lower concentration regime).
In general, the primary factor determining the spontaneity of addition is the concentration of pinic acid rather than the concentration of the FNP compounds. Increasing the pinic acid concentration from 0.5 to 20 ppt raises the maximum spontaneous condensation temperature from 221 to 236 K for the addition onto the SA–AM FNP and from 231 to 246 K for the addition onto the SA–MA FNP. Conversely, changing the base concentrations from the lower to the upper limits results in, at most, a 1 K difference. These findings suggest that pinic acid uptake is unlikely, even for the median concentration of 10 ppt, as it requires a temperature of 243 K for spontaneous addition onto the AM FNP. The addition to the MA FNP, however, is more favourable due to the direct proportionality between the addition temperature and the free energy change (Eq. 7). In the studied concentration range, the roughly 0.8 kcal mol−1 difference in addition free energy for the AM FNP compared to MA FNP results in a 10 K increase in the condensation temperature. Hence, the results are quite sensitive to the calculated free energies.
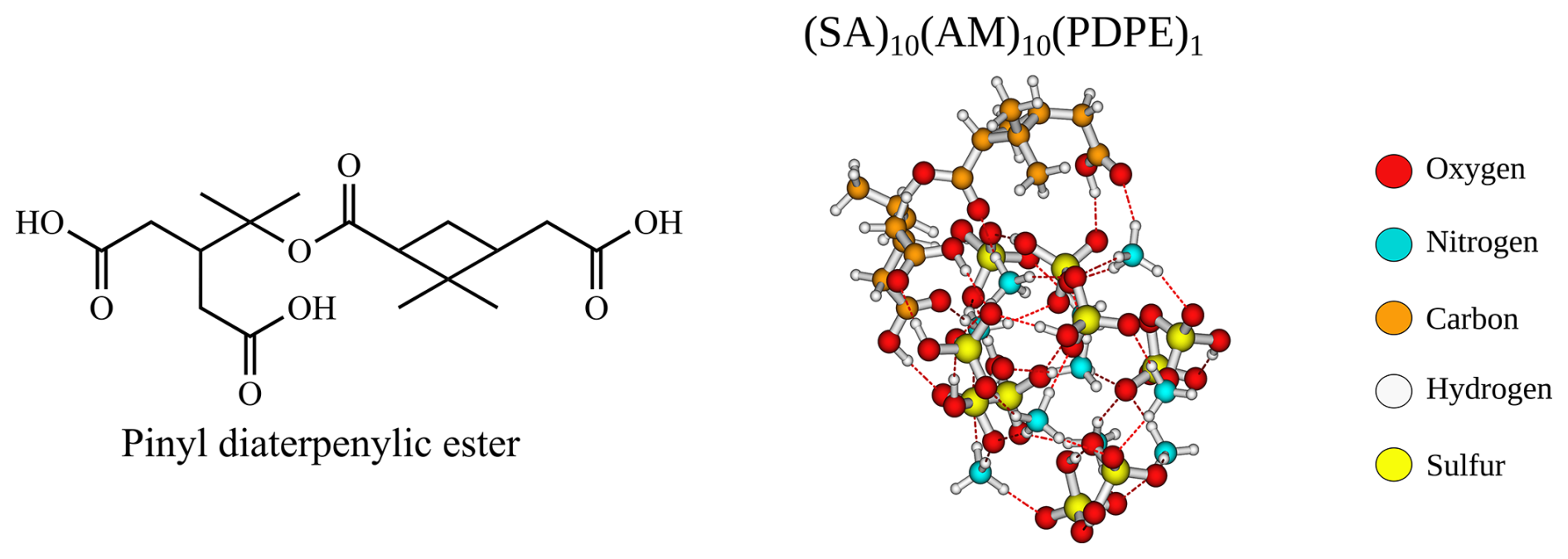
Figure 11The structure and lowest Gibbs free configuration of the (SA)10(AM)10(PDPE)1 FNP at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
The NA and FA uptake onto the (SA)10(AM)10 FNP stand out compared to the other monomers, as their higher atmospheric concentrations (ppb rather than ppt) may allow spontaneous uptake, despite their standard addition free energy exceeding −4 kcal mol−1. However, even at high concentrations (20.5 ppb) in the upper-concentration regime, a spontaneous addition of FA and NA requires temperatures below 209 and 174 K, respectively. This is due to the logarithmic dependence of the free energy on concentration (from Eq. 7), which minimises the temperature increase when shifting from ppt to ppb.
These temperatures are significantly colder than those reported in CLOUD chamber experiments by Wang et al. (2020) and Wang et al. (2022), where upper-tropospheric conditions were sufficient for NA condensation. However, their experiments measured particles larger than 2 nm, whereas our SA–AM FNP is only 1.4 nm. These results suggest that monomer uptake cannot overcome the Kelvin effect for the studied FNP sizes under realistic conditions. Nonetheless, pinic acid remains the most promising candidate for spontaneous condensation onto larger FNPs under cold, but plausible, atmospheric conditions.
Another way to increase the probability of uptake is to increase the number of interacting groups in the uptake monomers. The cluster-of-functional-groups approach by Pedersen et al. (2024) states that the binding free energies of organics can, as a first approximation, be estimated by their functional groups. For uptake on the (SA)10(AM)10 FNP, an organic with the functional groups of pinic acid (−8.4 kcal mol−1) and pinonic acid (−10.5 kcal mol−1) would give roughly −19 kcal mol−1. The accretion product pinyl diaterpenylic ester (PDPE) (Kristensen et al., 2013, 2016) has three carboxylic groups and one ester group and should approximately function as pinonic and pinic acid combined (see Fig. 11). The addition free energy for PDPE on the (SA)10(AM)10 FNP is −17.1 kcal mol−1, and the structure is depicted in Fig. 11.
When calculating the free energy under given conditions in the upper-concentration regime of FNP constituents, we find that for a concentration of 0.3 ppt for PDPE, the uptake is spontaneous at 298.15 K. At 0.03 ppt, the uptake is spontaneous at 278.15 K. This suggests that the extremely low volatile accretion products can overcome the Kelvin effect for FNPs at these sizes, but the low-volatility monomer oxidation products cannot.
We have investigated the ability of (SA)10(base)10 freshly nucleated particles (FNPs) with SA = sulfuric acid and base = ammonia (AM), methylamine (MA), dimethylamine (DMA), and trimethylamine (TMA) to uptake first-generation oxidation products of isoprene (trans-β-IEPOX, β4-ISPOOH, and β1-ISOPOOH), α-pinene (pinic and pinonic acid), a potential highly oxidised molecule (HOM), formic acid (FA), and nitric acid (NA). This was done using quantum chemical methods at the ωB97X-D3BJ/6-311++G(3df,3pd)//B97-3c level of theory.
We find that the HOM and pinic and pinonic acid can exhibit large decreases in addition free energies between −8 and −10 kcal mol−1, making them potential candidates for the organic growth of FNPs. This suggests that the studied isoprene oxidation products do not contribute to the early growth of FNPs, but the α-pinene products do. To further investigate this, we calculated the second addition free energy of pinic and pinonic acid onto the (SA)10(AM)10 FNP. We find that the pinic acid still exhibits a large addition free energy decrease of −11.8 kcal mol−1, but the secondary addition of pinonic acid drops to −3.9 kcal mol−1. The reason pinic acid maintains its high addition free energy decrease is due to its two carboxylic acid groups. The functional groups enable the pinic acid monomer to bind not only to the FNP surface but also to adjacent pinic acid. Calculating the addition free energy under realistic atmospheric conditions, we find that the FNPs studied are too small to support the growth of the studied monomer compounds, and extremely low-volatility accretion products are required for spontaneous uptake on the studied FNPs.
In the future, we imagine that molecular-dynamics-based sampling techniques, such as umbrella sampling, can give better insight into the effects of multiple minima and the partitioning of the uptake or collision simulations using molecular dynamics for realistic collision coefficients. It would also be interesting to study whether organic compounds can influence the cluster-to-particle transition points.
All the calculated structures and thermochemistry are available in the Atmospheric Cluster Database (ACDB) at https://doi.org/10.1021/acsomega.9b00860 (Elm, 2019) and at https://doi.org/10.1021/acsomega.3c07412 (Kubečka et al., 2023a).
JE: conceptualisation, project administration, funding acquisition, supervision, and resources. YK: formal analysis, investigation, writing (original draft), and visualisation. YK and JE: methodology and writing (review and editing).
At least one of the (co-)authors is a member of the editorial board of Aerosol Research. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
The views and opinions expressed are, however, those of the authors only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The numerical results presented in this work were obtained at the Centre for Scientific Computing, Aarhus https://phys.au.dk/forskning/faciliteter/cscaa/ (last access: 21 February 2025).
This work has been funded by the European Union (ERC, ExploreFNP; project no. 101040353) and by the Danish National Research Foundation (DNRF172) through the Center of Excellence for Chemistry of Clouds.
This paper was edited by Jose Castillo and reviewed by two anonymous referees.
Almeida, J., Schobesberger, S., Kürten, A., et al.: Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013. a
Ayoubi, D., Knattrup, Y., and Elm, J.: Clusteromics V: Organic Enhanced Atmospheric Cluster Formation, ACS Omega, 8, 9621–9629, https://doi.org/10.1021/acsomega.3c00251, 2023. a, b
Bannwarth, C., Caldeweyher, E., Ehlert, S., Hansen, A., Pracht, P., Seibert, J., Spicher, S., and Grimme, S.: Extended Tight-Binding Quantum Chemistry Methods, WIREs Comput. Mol. Sci., 11, e1493, https://doi.org/10.1002/wcms.1493, 2021. a
Boucher, O. and Lohmann, U.: The Sulfate-CCN-Cloud Albedo Effect, Tellus B, 47, 281–300, https://doi.org/10.3402/tellusb.v47i3.16048, 1995. a, b, c
Brandenburg, J. G., Bannwarth, C., Hansen, A., and Grimme, S.: B97-3c: A Revised Low-Cost Variant of the B97-D Density Functional Method, J. Chem. Phys., 148, 064104, https://doi.org/10.1063/1.5012601, 2018. a
Bready, C. J., Fowler, V. R., Juechter, L. A., Kurfman, L. A., Mazaleski, G. E., and Shields, G. C.: The Driving Effects of Common Atmospheric Molecules for Formation of Prenucleation Clusters: The Case of Sulfuric acid, Formic acid, Nitric acid, Ammonia, and Dimethyl Amine, Environ. Sci.: Atmos., 2, 1469–1486, https://doi.org/10.1039/D2EA00087C, 2022. a, b
Canadell, J. G., Monteiro, P. M. S., Costa, M. H., et al.: 5 - Global Carbon and other Biogeochemical Cycles and Feedbacks, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 673–816, https://doi.org/10.1017/9781009157896.007, 2021. a, b
DePalma, J. W., Wang, J., Wexler, A. S., and Johnston, M. V.: Growth of Ammonium Bisulfate Clusters by Adsorption of Oxygenated Organic Molecules, J. Phys. Chem. A, 119, 11191–11198, https://doi.org/10.1021/acs.jpca.5b07744, 2015. a
Ditchfield, R., Hehre, W. J., and Pople, J. A.: Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules, J. Chem. Phys., 54, 724–728, https://doi.org/10.1063/1.1674902, 1971. a
Dunne, E. M., Gordon, H., Kürten, A., et al.: Global Atmospheric Particle Formation from CERN CLOUD Measurements, Science, 354, 1119–1124, https://doi.org/10.1126/science.aaf2649, 2016. a
Elm, J.: An Atmospheric Cluster Database Consisting of Sulfuric Acid, Bases, Organics, and Water, ACS Omega, 4, 10965–10974, https://doi.org/10.1021/acsomega.9b00860, 2019. a, b
Elm, J.: Clusteromics I: Principles, Protocols, and Applications to Sulfuric Acid–Base Cluster Formation, ACS Omega, 6, 7804–7814, https://doi.org/10.1021/acsomega.1c00306, 2021a. a, b, c, d
Elm, J.: Clusteromics II: Methanesulfonic Acid–Base Cluster Formation, ACS Omega, 6, 17035–17044, https://doi.org/10.1021/acsomega.1c02115, 2021b. a
Elm, J.: Clusteromics III: Acid Synergy in Sulfuric Acid–Methanesulfonic Acid–Base Cluster Formation, ACS Omega, 7, 15206–15214, https://doi.org/10.1021/acsomega.2c01396, 2022. a
Elm, J., Myllys, N., and Kurtén, T.: What Is Required for Highly Oxidized Molecules To Form Clusters with Sulfuric Acid?, J. Phys. Chem. A, 121, 4578–4587, https://doi.org/10.1021/acs.jpca.7b03759, 2017. a
Elm, J., Kubečka, J., Besel, V., Jääskeläinen, M. J., Halonen, R., Kurtén, T., and Vehkamäki, H.: Modeling the Formation and Growth of Atmospheric Molecular Clusters: A Review, J. Aerosol Sci., 149, 105621, https://doi.org/10.1016/j.jaerosci.2020.105621, 2020. a, b
Elm, J., Ayoubi, D., Engsvang, M., Jensen, A. B., Knattrup, Y., Kubečka, J., Bready, C. J., Fowler, V. R., Harold, S. E., Longsworth, O. M., and Shields, G. C.: Quantum Chemical Modeling of Organic Enhanced Atmospheric Nucleation: A Critical Review, WIREs Comput. Mol. Sci., 13, e1662, https://doi.org/10.1002/wcms.1662, 2023. a, b
Engsvang, M. and Elm, J.: Modeling the Binding Free Energy of Large Atmospheric Sulfuric Acid–Ammonia Clusters, ACS Omega, 7, 8077–8083, https://doi.org/10.1021/acsomega.1c07303, 2022. a
Engsvang, M., Kubečka, J., and Elm, J.: Toward Modeling the Growth of Large Atmospheric Sulfuric Acid–Ammonia Clusters, ACS Omega, 8, 34597–34609, https://doi.org/10.1021/acsomega.3c03521, 2023a. a, b, c, d
Engsvang, M., Wu, H., Knattrup, Y., Kubečka, J., Jensen, A. B., and Elm, J.: Quantum Chemical Modeling of Atmospheric Molecular Clusters Involving Inorganic Acids and Methanesulfonic Acid, Chem. Phys. Rev., 4, 031311, https://doi.org/10.1063/5.0152517, 2023b. a, b
Gaussian, A., Frisch, M. J., Trucks, G. W., et al.: Gaussian 16, Rev.B.01, Gaussian, Inc., Wallingford CT, 2016. a
Glasoe, W. A., Volz, K., Panta, B., Freshour, N., Bachman, R., Hanson, D. R., McMurry, P. H., and Jen, C.: Sulfuric acid Nucleation: An Experimental Study of the Effect of Seven Bases, J. Geophys. Res.-Atmos., 120, 1933–1950, https://doi.org/10.1002/2014JD022730, 2015. a, b, c
Grimme, S.: Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory, Chem. Eur. J., 18, 9955–9964, https://doi.org/10.1002/chem.201200497, 2012. a
Grimme, S.: Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations, J. Chem. Theory Comput., 15, 2847–2862, https://doi.org/10.1021/acs.jctc.9b00143, 2019. a
Grimme, S., Bannwarth, C., and Shushkov, P.: A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86), J. Chem. Theory Comput., 13, 1989–2009, https://doi.org/10.1021/acs.jctc.7b00118, 2017. a
Halonen, R.: A Consistent Formation Free Energy Definition for Multicomponent Clusters in Quantum Thermochemistry, J. Aerosol Sci., 162, 105974, https://doi.org/10.1016/j.jaerosci.2022.105974, 2022. a
Harold, S. E., Bready, C. J., Juechter, L. A., Kurfman, L. A., Vanovac, S., Fowler, V. R., Mazaleski, G. E., Odbadrakh, T. T., and Shields, G. C.: Hydrogen-Bond Topology Is More Important Than Acid/Base Strength in Atmospheric Prenucleation Clusters, J. Phys. Chem. A, 126, 1718–1728, https://doi.org/10.1021/acs.jpca.1c10754, 2022. a
Hasan, G., Wu, H., Knattrup, Y., and Elm, J.: Base synergy in freshly nucleated particles, Aerosol Research Discuss. [preprint], https://doi.org/10.5194/ar-2024-28, in review, 2024. a
Hunter, E. P. L. and Lias, S. G.: Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. Chem. Ref. Data, 27, 413–656, https://doi.org/10.1063/1.556018, 1998. a
Jen, C. N., McMurry, P. H., and Hanson, D. R.: Stabilization of Sulfuric Acid Dimers by Ammonia, Methylamine, Dimethylamine, and Trimethylamine, J. Geophys. Res.-Atmos., 119, 7502–7514, https://doi.org/10.1002/2014JD021592, 2014. a, b, c
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012. a
Kirkby, J., Curtius, J., Almeida, J., et al.: Role of Sulphuric Acid, Ammonia and Galactic Cosmic Rays in Atmospheric Aerosol Nucleation, Nature, 476, 429–433, https://doi.org/10.1038/nature10343, 2011. a
Knattrup, Y. and Elm, J.: Clusteromics IV: The Role of Nitric Acid in Atmospheric Cluster Formation, ACS Omega, 7, 31551–31560, https://doi.org/10.1021/acsomega.2c04278, 2022. a, b
Knattrup, Y., Kubečka, J., and Elm, J.: Nitric Acid and Organic Acids Suppress the Role of Methanesulfonic Acid in Atmospheric New Particle Formation, J. Phys. Chem. A, 127, 7568–7578, https://doi.org/10.1021/acs.jpca.3c04393, 2023. a, b
Knattrup, Y., Kubečka, J., Wu, H., Jensen, F., and Elm, J.: Reparameterization of GFN1-xTB for Atmospheric Molecular Clusters: Applications to Multi-Acid–Multi-Base Systems, RSC Adv., 14, 20 048–20 055, https://doi.org/10.1039/D4RA03021D, 2024. a, b
Kristensen, K., Enggrob, K. L., King, S. M., Worton, D. R., Platt, S. M., Mortensen, R., Rosenoern, T., Surratt, J. D., Bilde, M., Goldstein, A. H., and Glasius, M.: Formation and occurrence of dimer esters of pinene oxidation products in atmospheric aerosols, Atmos. Chem. Phys., 13, 3763–3776, https://doi.org/10.5194/acp-13-3763-2013, 2013. a
Kristensen, K., Watne, Å. K., Hammes, J., Lutz, A., Petäjä, T., Hallquist, M., Bilde, M., and Glasius, M.: High-Molecular Weight Dimer Esters Are Major Products in Aerosols from α-Pinene Ozonolysis and the Boreal Forest, Environ. Sci. Technol. Lett., 3, 280–285, https://doi.org/10.1021/acs.estlett.6b00152, 2016. a
Kubečka, J., Besel, V., Neefjes, I., Knattrup, Y., Kurtén, T., Vehkamäki, H., and Elm, J.: Computational Tools for Handling Molecular Clusters: Configurational Sampling, Storage, Analysis, and Machine Learning, ACS Omega, 8, 45115–45128, https://doi.org/10.1021/acsomega.3c07412, 2023a. a, b, c
Kubečka, J., Neefjes, I., Besel, V., Qiao, F., Xie, H.-B., and Elm, J.: Atmospheric Sulfuric Acid–Multi-Base New Particle Formation Revealed through Quantum Chemistry Enhanced by Machine Learning, J. Phys. Chem. A, 27, 2091–2103, https://doi.org/10.1021/acs.jpca.3c00068, 2023b. a, b, c, d
Kulmala, M., Kontkanen, J., Junninen, H., Lehtipalo, K., Manninen, H. E., Nieminen, T., Petäjä, T., Sipilä, M., Schobesberger, S., Rantala, P., Franchin, A., Jokinen, T., Järvinen, E., Äijälä, M., Kangasluoma, J., Hakala, J., Aalto, P. P., Paasonen, P., Mikkilä, J., Vanhanen, J., Aalto, J., Hakola, H., Makkonen, U., Ruuskanen, T., Mauldin, R. L., Duplissy, J., Vehkamäki, H., Bäck, J., Kortelainen, A., Riipinen, I., Kurtén, T., Johnston, M. V., Smith, J. N., Ehn, M., Mentel, T. F., Lehtinen, K. E. J., Laaksonen, A., Kerminen, V.-M., and Worsnop, D. R.: Direct Observations of Atmospheric Aerosol Nucleation, Science, 339, 943–946, https://doi.org/10.1126/science.1227385, 2013. a, b, c
Kumar, M., Li, H., Zhang, X., Zeng, X. C., and Francisco, J. S.: Nitric Acid–Amine Chemistry in the Gas Phase and at the Air–Water Interface, J. Am. Chem. Soc., 140, 6456–6466, https://doi.org/10.1021/jacs.8b03300, 2018. a
Kurtén, T., Loukonen, V., Vehkamäki, H., and Kulmala, M.: Amines are likely to enhance neutral and ion-induced sulfuric acid-water nucleation in the atmosphere more effectively than ammonia, Atmos. Chem. Phys., 8, 4095–4103, https://doi.org/10.5194/acp-8-4095-2008, 2008. a, b, c
Kurtén, T., Tiusanen, K., Roldin, P., Rissanen, M., Luy, J.-N., Boy, M., Ehn, M., and Donahue, N.: α-Pinene Autoxidation Products May Not Have Extremely Low Saturation Vapor Pressures Despite High O:C Ratios, J. Phys. Chem. A, 120, 2569–2582, https://doi.org/10.1021/acs.jpca.6b02196, 2016. a
Ling, J., Ding, X., Li, Z., and Yang, J.: First-Principles Study of Molecular Clusters Formed by Nitric Acid and Ammonia, J. Phys. Chem. A, 121, 661–668, https://doi.org/10.1021/acs.jpca.6b09185, 2017. a
Liu, L., Li, H., Zhang, H., Zhong, J., Bai, Y., Ge, M., Li, Z., Chen, Y., and Zhang, X.: The role of Nitric Acid in Atmospheric New Particle Formation, Phys. Chem. Chem. Phys., 20, 17406–17414, https://doi.org/10.1039/C8CP02719F, 2018. a
Liu, L., Yu, F., Du, L., Yang, Z., Francisco, J. S., and Zhang, X.: Rapid Sulfuric Acid–Dimethylamine Nucleation Enhanced by Nitric Acid in Polluted Regions, P. Natl. Acad. Sci. USA, 118, e2108384118, https://doi.org/10.1073/pnas.2108384118, 2021. a
Longsworth, O. M., Bready, C. J., Joines, M. S., and Shields, G. C.: The Driving Effects of Common Atmospheric Molecules for Formation of Clusters: The Case of Sulfuric Acid, Nitric Acid, Hydrochloric Acid, Ammonia, and Dimethylamine, Environ. Sci.: Atmos., 3, 1585–1600, https://doi.org/10.1039/D3EA00118K, 2023. a
Loukonen, V., Kurtén, T., Ortega, I. K., Vehkamäki, H., Pádua, A. A. H., Sellegri, K., and Kulmala, M.: Enhancing effect of dimethylamine in sulfuric acid nucleation in the presence of water – a computational study, Atmos. Chem. Phys., 10, 4961–4974, https://doi.org/10.5194/acp-10-4961-2010, 2010. a
Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J., and Carslaw, K. S.: Impact of nucleation on global CCN, Atmos. Chem. Phys., 9, 8601–8616, https://doi.org/10.5194/acp-9-8601-2009, 2009. a
Nadykto, A. B. and Yu, F.: Strong Hydrogen Bonding between Atmospheric Nucleation Precursors and common Organics, Chem. Phys. Lett., 435, 14–18, https://doi.org/10.1016/j.cplett.2006.12.050, 2007. a
Nadykto, A. B., Yu, F., Jakovleva, M. V., Herb, J., and Xu, Y.: Amines in the Earth's Atmosphere: A Density Functional Theory Study of the Thermochemistry of Pre-Nucleation Clusters, Entropy, 13, 554–569, https://doi.org/10.3390/e13020554, 2011. a, b, c
Nadykto, A. B., Herb, J., Yu, F., and Xu, Y.: Enhancement in the Production of Nucleating Clusters due to Dimethylamine and Large Uncertainties in the Thermochemistry of Amine-Enhanced Nucleation, Chem. Phys. Lett., 609, 42–49, https://doi.org/10.1016/j.cplett.2014.03.036, 2014. a, b
Nadykto, A. B., Herb, J., Yu, F., Xu, Y., and Nazarenko, E. S.: Estimating the Lower Limit of the Impact of Amines on Nucleation in the Earth's Atmosphere, Entropy, 17, 2764–2780, https://doi.org/10.3390/e17052764, 2015. a, b, c
Najibi, A. and Goerigk, L.: The Nonlocal Kernel in van der Waals Density Functionals as an Additive Correction: An Extensive Analysis with Special Emphasis on the B97M-V and ωB97M-V Approaches, J. Chem. Theory Comput., 14, 5725–5738, https://doi.org/10.1021/acs.jctc.8b00842, 2018. a
Neese, F.: The ORCA Program System, WIREs Comput. Mol. Sci., 2, 73–78, https://doi.org/10.1002/wcms.81, 2012. a
Neese, F.: Software Update: The ORCA Program System – Version 5.0, WIREs Comput. Mol. Sci., 12, e1606, https://doi.org/10.1002/wcms.1606, 2022. a
Neese, F., Wennmohs, F., Becker, U., and Riplinger, C.: The ORCA Quantum Chemistry Program Package, J. Chem. Phys., 152, 224108, https://doi.org/10.1063/5.0004608, 2020. a
Nguyen, M.-T., Jamka, A. J., Cazar, R. A., and Tao, F.-M.: Structure and Stability of the Nitric Acid–Ammonia Complex in the Gas Phase and in Water, J. Chem. Phys., 106, 8710–8717, https://doi.org/10.1063/1.473925, 1997. a
Nozière, B., Kalberer, M., Claeys, M., Allan, J., D'Anna, B., Decesari, S., Finessi, E., Glasius, M., Grgić, I., Hamilton, J. F., Hoffmann, T., Iinuma, Y., Jaoui, M., Kahnt, A., Kampf, C. J., Kourtchev, I., Maenhaut, W., Marsden, N., Saarikoski, S., Schnelle-Kreis, J., Surratt, J. D., Szidat, S., Szmigielski, R., and Wisthaler, A.: The Molecular Identification of Organic Compounds in the Atmosphere: State of the Art and Challenges, Chem. Rev., 115, 3919–3983, https://doi.org/10.1021/cr5003485, 2015. a
Passananti, M., Zapadinsky, E., Zanca, T., Kangasluoma, J., Myllys, N., Rissanen, M. P., Kurtén, T., Ehn, M., Attoui, M., and Vehkamäki, H.: How Well can we Predict Cluster Fragmentation inside a Mass Spectrometer?, Chem. Commun., 55, 5946–5949, https://doi.org/10.1039/C9CC02896J, 2019. a
Pedersen, A. N., Knattrup, Y., and Elm, J.: A cluster-of-functional-groups approach for studying organic enhanced atmospheric cluster formation, Aerosol Research, 2, 123–134, https://doi.org/10.5194/ar-2-123-2024, 2024. a, b
Pracht, P., Bohle, F., and Grimme, S.: Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods, Phys. Chem. Chem. Phys., 22, 7169–7192, https://doi.org/10.1039/C9CP06869D, 2020. a
Qiao, F., Zhang, R., Zhao, Q., Ma, F., Chen, J., and Xie, H.-B.: A Surprisingly High Enhancing Potential of Nitric Acid in Sulfuric Acid–Methylamine Nucleation, Atmosphere, 15, 467, https://doi.org/10.3390/atmos15040467, 2024. a
Schobesberger, S., Junninen, H., Bianchi, F., Lönn, G., Ehn, M., Lehtipalo, K., Dommen, J., Ehrhart, S., Ortega, I. K., Franchin, A., Nieminen, T., Riccobono, F., Hutterli, M., Duplissy, J., Almeida, J., Amorim, A., Breitenlechner, M., Downard, A. J., Dunne, E. M., Flagan, R. C., Kajos, M., Keskinen, H., Kirkby, J., Kupc, A., Kürten, A., Kurtén, T., Laaksonen, A., Mathot, S., Onnela, A., Praplan, A. P. Rondo, L., Santos, F. D. Schallhart, S., Schnitzhofer, R., Sipilä, M., Tomé, A., Tsagkogeorgas, G., Vehkamäki, H., Wimmer, D., Baltensperger, U., Carslaw, K. S., Curtius, J., Hansel, A., Petäjä, T., Kulmala, M., Donahue, N. M., and Worsnop, D. R.: Molecular Understanding of Atmospheric Particle Formation from Sulfuric Acid and Large Oxidized Organic Molecules, P. Natl. Acad. Sci. USA, 110, 17223–17228, https://doi.org/10.1073/pnas.1306973110, 2013. a
Stolzenburg, D., Fischer, L., Vogel, A. L., et al.: Rapid Growth of Organic Aerosol Nanoparticles over a Wide Tropospheric Temperature Range, P. Natl. Acad. Sci. USA, 115, 9122–9127, https://doi.org/10.1073/pnas.1807604115, 2018. a
Tröstl, J., Chuang, W. K., Gordon, H., et al.: The Role of Low-Volatility Organic Compounds in Initial Particle Growth in the Atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016. a
Wang, M., Kong, W., Marten, R., et al.: Rapid Growth of New Atmospheric Particles by Nitric Acid and Ammonia Condensation, Nature, 581, 184–189, https://doi.org/10.1038/s41586-020-2270-4, 2020. a, b, c
Wang, M., Xiao, M., Bertozzi, B., Marie, G., et al.: Synergistic HNO3–H2SO4–NH3 Upper Tropospheric Particle Formation, Nature, 605, 483–489, https://doi.org/10.1038/s41586-022-04605-4, 2022. a, b, c
Weber, R. J., Marti, J. J., Mcmurry, P. H., Eisele, F. L., Tanner, D. J., and Jefferson, A.: Measured Atmospheric New Particle Formation Rates: Implications for Nucleation Mechanisms, Chem. Eng. Commun., 151, 53–64, https://doi.org/10.1080/00986449608936541, 1996. a
Wu, H., Engsvang, M., Knattrup, Y., Kubečka, J., and Elm, J.: Improved Configurational Sampling Protocol for Large Atmospheric Molecular Clusters, ACS Omega, 8, 45065–45077, https://doi.org/10.1021/acsomega.3c06794, 2023. a, b
Wu, H., Knattrup, Y., Jensen, A. B., and Elm, J.: Cluster-to-particle transition in atmospheric nanoclusters, Aerosol Research, 2, 303–314, https://doi.org/10.5194/ar-2-303-2024, 2024. a, b, c, d, e, f, g
Zapadinsky, E., Passananti, M., Myllys, N., Kurtén, T., and Vehkamäki, H.: Modeling on Fragmentation of Clusters inside a Mass Spectrometer, J. Phys. Chem. A, 123, 611–624, https://doi.org/10.1021/acs.jpca.8b10744, 2019. a
Zhang, J.: Atom Typing using Graph Representation Learning: How do Models Learn Chemistry?, J. Chem. Phys., 156, 204108, https://doi.org/10.1063/5.0095008, 2022. a, b
Zhang, J. and Dolg, M.: ABCluster: The Artificial Bee Colony Algorithm for Cluster Global Optimization, Phys. Chem. Chem. Phys., 17, 24173–24181, https://doi.org/10.1039/C5CP04060D, 2015. a, b, c
Zhang, J. and Dolg, M.: Global Optimization of Clusters of Rigid Molecules using the Artificial Bee Colony Algorithm, Phys. Chem. Chem. Phys., 18, 3003–3010, https://doi.org/10.1039/C5CP06313B, 2016. a, b, c
Zhang, R., Jiang, S., Liu, Y.-R., Wen, H., Feng, Y.-J., Huang, T., and Huang, W.: An Investigation about the Structures, Thermodynamics and Kinetics of the Formic Acid involved Molecular Clusters, Chem. Phys., 507, 44–50, https://doi.org/10.1016/j.chemphys.2018.03.029, 2018. a
Zhang, R., Shen, J., Xie, H.-B., Chen, J., and Elm, J.: The role of organic acids in new particle formation from methanesulfonic acid and methylamine, Atmos. Chem. Phys., 22, 2639–2650, https://doi.org/10.5194/acp-22-2639-2022, 2022. a
Zhao, B., Donahue, N. M., Zhang, K., Mao, L., Shrivastava, M., Ma, P.-L., Shen, J., Wang, S., Sun, J., Gordon, H., Tang, S., Fast, J., Wang, M., Gao, Y., Yan, C., Singh, B., Li, Z., Huang, L., Lou, S., Lin, G., Wang, H., Jiang, J., Ding, A., Nie, W., Qi, X., Chi, X., and Wang, L.: Global Variability in Atmospheric New Particle Formation mechanisms, Nature, 631, 98–105, https://doi.org/10.1038/s41586-024-07547-1, 2024. a