the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Apr 2025
| 03 Apr 2025
The impact of unimolecular reactions on acyl peroxy radical initiated isoprene oxidation
Ida Karppinen
Dominika Pasik
Emelda Ahongshangbam
Unimolecular H-shift and endoperoxide ring formation reactions were studied for several different acyl peroxy radicals (APRs) using quantum-mechanical methods. Also, for structures with slow unimolecular reactions, accretion reactions with isoprene were investigated. The goal of the study was to determine which APRs could work as atmospheric oxidants of unsaturated hydrocarbons. The reaction rate coefficients were calculated at the DLPNO-CCSD(T)/aug-cc-pVTZ//ωB97X-D/6-31+G* level using multi-conformer transition state theory. Unimolecular reactions of acyl peroxy radicals were shown to have rate coefficients of up to 0.1 s−1 and bimolecular accretion reactions with isoprene of up to 10−15 cm3 s−1. Both smaller and larger acyl peroxy radicals with rigid structures were observed to be more likely to initiate oxidation of isoprene because of their inability in fast unimolecular reactions. The pseudo-first-order reaction rates were calculated for accretion reactions of isoprene with OH and six APRs at different temperatures. The significance of APR-initiated isoprene oxidation was shown to increase with increasing temperature. APR-initiated oxidation could lead to dimeric products with atmospheric impact through formation of low-volatility compounds.
- Article
(843 KB) - Full-text XML
-
Supplement
(686 KB) - BibTeX
- EndNote




Peroxy radicals (RO2) can undergo a variety of different reactions in the atmosphere. RO2 reactions with NOx and NO3 are important under moderately to highly polluted conditions. However, in clean environments, reactions with other species, mainly HO2, become more significant (Jenkin et al., 2019). Unimolecular reactions, such as H-shift reactions and endoperoxide ring formation reactions, are also possible reaction pathways for RO2 radicals. An interesting class of RO2 radicals is acyl peroxy radicals (APRs), which have proven to be more reactive than other RO2 radicals (Knap and Jørgensen, 2017; Seal et al., 2023; Møller et al., 2019; Vereecken and Nozière, 2020; Nozière and Fache, 2021). APRs are generally formed from aldehydes when the aldehydic hydrogen atom is removed by photolysis or in a reaction with the OH radical (Demore et al., 1997; Atkinson et al., 1997). In these reactions, an acyl radical forms and rapidly reacts with O2, forming an APR (Atkinson and Arey, 2003). APRs can also be formed by photolysis of ketones (El-Agamey and McGarvey, 2002) or indirectly from aldehydes through a process called autoxidation (Crounse et al., 2013; Knap and Jørgensen, 2017). However, the direct APR formation route from aldehydes is the dominant one, since the removal of an aldehydic H atom is much faster than the removal of a non-aldehydic H atom (Calvert et al., 2011).
Studies have shown that APRs, unlike other RO2 radicals, are reactive enough towards double bonds for these reactions to occur under atmospheric conditions (Nozière and Fache, 2021; Pasik et al., 2024a). Experimental studies of RO2 gas-phase reactions with alkenes at high temperatures have also shown that APRs are more reactive towards double bonds than other RO2 (Stark, 1997). Accretion reaction of an APR with an unsaturated hydrocarbon leads to a dimeric product with an alkyl radical center (Nozière et al., 2023). O2 can add to this radical center, forming new RO2 radicals which may undergo autoxidation. The described APR-initiated oxidation leads to compounds with high molecular mass and multiple oxygen atoms. These compounds have low vapor pressure and therefore can potentially participate in atmospheric new particle formation (NPF). NPF accounts for a major part of tropospheric aerosol production, which can act as cloud condensation nuclei (CCN) and negatively affect human health (Lee et al., 2019). However, it has also been found that APRs may go through fast H-shift reactions (Knap and Jørgensen, 2017; Møller et al., 2019; Vereecken and Nozière, 2020; Seal et al., 2023), making their reactions with unsaturated hydrocarbons less likely. Also, APRs with a double bond in their structure can go through endoperoxide ring formation reactions (Vereecken et al., 2021; Nozière and Vereecken, 2024). These reactions have been studied for RO2 radicals and have been shown to compete with H-shift reactions (Vereecken and Peeters, 2004; Xu et al., 2019; Møller et al., 2020). Vereecken et al. (2021) also studied some APR ring closure reactions and showed that APRs exhibit faster ring closure reactions compared to RO2 radicals.
The unimolecular H-shift and ring closure reactions of APRs lead to the formation of an alkyl radical to which O2 is rapidly added, forming a peroxy radical. This means that if APR undergoes fast unimolecular reactions, it cannot react with unsaturated hydrocarbons. Therefore, these unimolecular reactions have to be slow enough to allow accretion reactions to double-bond, which has been illustrated in Fig. 1. Previous studies on unimolecular reactions of APRs have mainly focused on which of these reactions are fast under atmospheric conditions. The aim of this study is to find APR structures with slow ( s−1) unimolecular reactions but fast ( s−1) bimolecular accretion reactions to double-bond. Our goal is to determine which types of APRs could work as oxidants of unsaturated hydrocarbons and initiate the formation of low-volatility compounds. These APRs with slow unimolecular reactions and fast bimolecular accretion reactions to double-bond are potential oxidants of unsaturated hydrocarbons. Hydroxyl radical (OH) accounts for 66 %–95 % of isoprene oxidation (Gu et al., 2022), and therefore the APR-initiated oxidation rate is compared to that of OH. Moreover, the temperature dependence of the bimolecular reactions is investigated, and the pseudo-first-order reaction rate coefficients are presented at different temperatures.
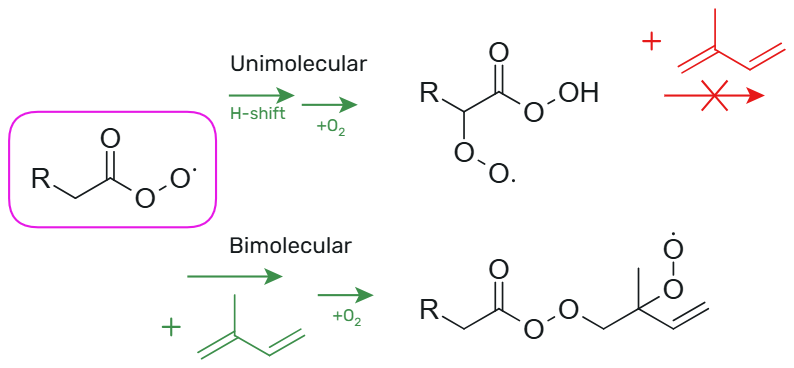
2.1 Computational details
Transition state (TS) structures were obtained using a relaxed potential energy surface (PES) scan and density functional theory (DFT) level ωB97X-D/6-31+G* (Chai and Head-Gordon, 2008b, a; Hehre et al., 1972; Clark et al., 1983). For unimolecular reactions, TS structures were located starting from the reactant, whereas for bimolecular reactions they were located starting from the product. Prior to the PES scan, the reactant and product structures were optimized at the ωB97X-D/6-31+G* level of theory. Reactant and TS conformers were found using the CREST sampling tool at the GFN2-xTB level (Pracht et al., 2020; Bannwarth et al., 2019). TS conformer configurational search included constraining the bonds forming the TS structure. For unimolecular H-shift reactions, this involved constraining the C−H and H−O bond distances, while for endoperoxide ring formation reactions the O−C bond being formed was constrained. For bimolecular reactions the C−O bond being broken was kept constrained. Reactant and TS conformers were optimized at the same DFT level as before using a 2.5 kcal mol−1 cutoff after the CREST configurational search, as xTB level electronic energies correlate well with DFT energies (Pasik et al., 2024b; Kubečka et al., 2019). Duplicates were removed based on electronic energy and dipole moments, and TS structures were confirmed by one imaginary frequency. For the lowest energy reactant and TS structures, single point energies were calculated using the DLPNO-CCSD(T)/aug-cc-pVTZ level of theory (Riplinger and Neese, 2013; Riplinger et al., 2013; Dunning, 1989; Kendall et al., 1992). All DFT calculations were carried out using the Gaussian 16 software, and ORCA version 5.0.3 was used for the single point energy calculations (Frisch et al., 2016; Neese, 2012).
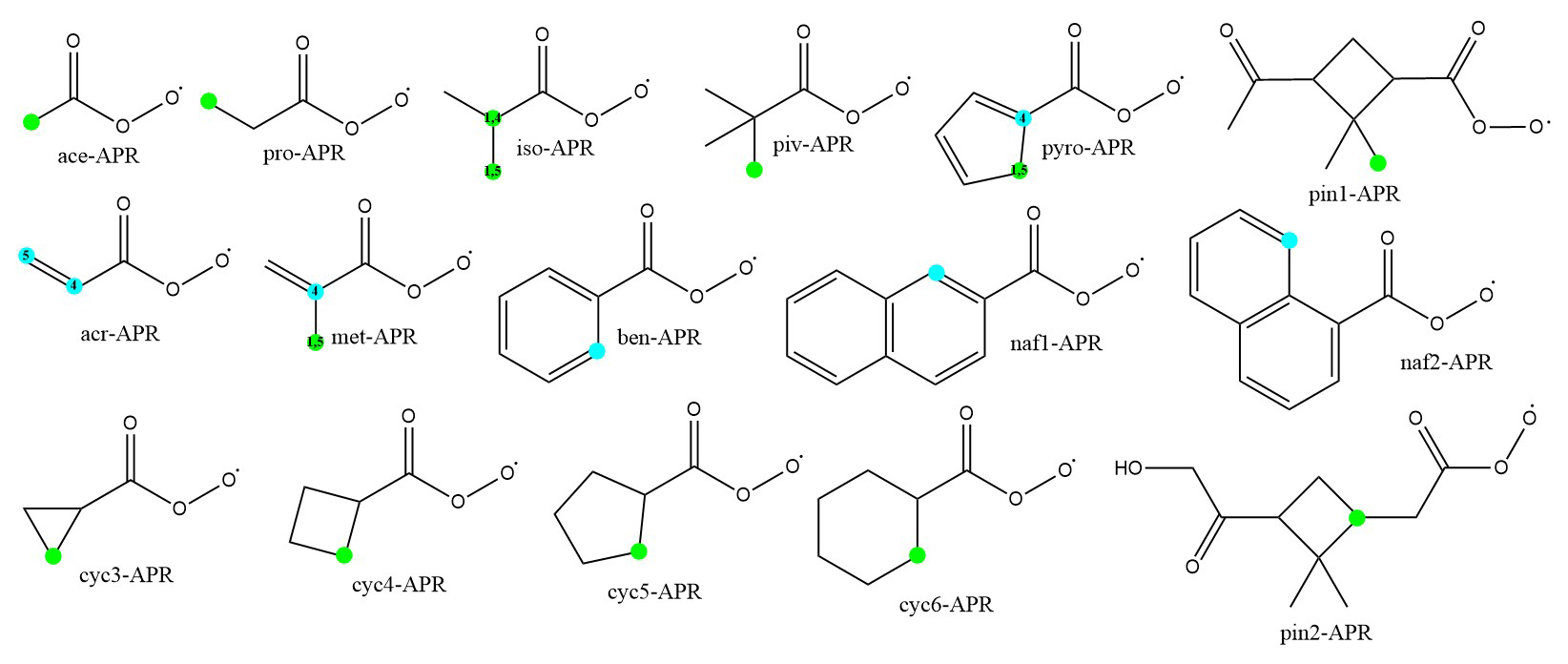
Figure 2The chemical structures of acyl peroxy radicals investigated in this study. The fastest unimolecular reactions are marked with dots, H-shifts in green, and endoperoxide ring formations in blue. For clarification, the prefixes for structures with two reactions with similar rates are also marked.
To compare the significance of APR-initiated isoprene oxidation to that of OH, we performed calculations for the reactions between isoprene and OH as well. The TS structures for the bimolecular reactions between OH and isoprene could not be found with the ωB97X-D functional. This is because the saddle point is very shallow or might not even exist (see Figs. S2 and S3 in the Supplement for the PES graphs). In addition, the pre-reactive complex that forms in the reaction is close in energy to the TS structure, further explaining why the TS is difficult to locate (see Fig. S4 for the energy diagrams). Thus, alternatively, the M06-2X functional was used (Zhao and Truhlar, 2008), and all reactant and TS conformers were optimized at the M06-2X/6-31+G* level for the reactions between OH and isoprene. On top of that, single point energies were calculated for the lowest energy conformers using the DLPNO-CCSD(T)/aug-cc-pVTZ level of theory. Three of the studied bimolecular reactions between isoprene and APRs (ace-APR reaction R4, pro-APR reaction R1, and ben-APR reaction R1) were additionally investigated at the DLPNO//M06-2X level to examine how the rate coefficients differ from those calculated at the DLPNO//ωB97X-D level. There is only a minor difference between zero-point corrected electronic energies at the studied DLPNO//DFT levels. However, partition functions between those density functionals differ significantly, which can affect an order of magnitude difference in the rate coefficients. The results from these benchmark calculations are provided in Table S2 in the Supplement.
2.2 Rate coefficients
Multi-conformer transition state theory (MC-TST) was utilized to calculate the reaction rate coefficients (Vereecken and Peeters, 2003). The rate coefficients for the unimolecular reactions were calculated using Eq. (1) (Møller et al., 2016):
where kB is Boltzmann's constant, T is the temperature, and h is Planck's constant. ΔEi is the zero-point corrected energy of TS conformer i relative to the lowest energy TS conformer and QTS,i is the partition function of TS conformer i, both of which are calculated at the ωB97X-D/6-31+G* level. ΔEj and QR,j are the analogous values for the reactant conformers. ETS and ER are the zero-point corrected energies of the lowest energy TS and the reactant conformer, respectively, including the DLPNO-CCSD(T)/aug-cc-pVTZ correction.
κt is the quantum-mechanical tunneling coefficient which was calculated using the one-dimensional Eckart tunneling method (Eckart, 1930; Johnston and Heicklen, 1962). Tunneling was needed for H-shift reactions due to the low mass of the hydrogen atom (McMahon, 2003). To calculate the tunneling coefficient, forward and reverse intrinsic reaction coordinate (IRC) calculations were carried out to connect the lowest energy TS to the corresponding reactant and product (Møller et al., 2016). The resulting reactant and product structures were optimized at the ωB97X-D/6-31+G* level, followed by DLPNO-CCSD(T)/aug-cc-pVTZ energy corrections. Additionally, the imaginary frequency of the lowest energy TS was utilized to calculate the tunneling coefficient.
The rate coefficients for the bimolecular reactions were calculated using Eq. (2) (Viegas, 2018, 2021):
where Pref is the reference pressure (2.45×1019 molecules per cubic centimeter) and Qip is the partition function of isoprene. The second isoprene conformer is 1.5 kcal mol−1 higher in energy than the lowest energy conformer, which would lead to a small exponential factor of 0.08 in Eq. (2) and a small contribution to the overall rate. Therefore, only the partition function of the lowest energy conformer of isoprene was considered in Eq. (2).
The rates of unimolecular H-shift and endoperoxide ring formation reactions for several different APR structures are calculated. In addition, for structures exhibiting slow unimolecular reaction rates, bimolecular accretion reactions with isoprene are investigated. The APR structures and their corresponding names used in this study are presented in Fig. 2. These structures include not only small APRs, but also larger cyclic APR structures that are potentially too rigid for fast unimolecular reactions.
3.1 Unimolecular reactions
We calculated the reaction rate coefficients for unimolecular H-shift and endoperoxide ring formation reactions. For clarity, Table 1 presents only the results for the fastest calculated rate coefficients. The fastest reactions are also illustrated in Fig. 2. Other calculated rate coefficients can be found in Table S1.
Table 1Calculated energy barrier heights (ΔETS; kcal mol−1), Eckart tunneling coefficients (κt) for H-shifts, and unimolecular MC-TST reaction rate coefficients (kuni; s−1) at 298 K of the fastest unimolecular reactions for the studied APRs. The APRs that have slow ( s−1) unimolecular reactions are in bold.
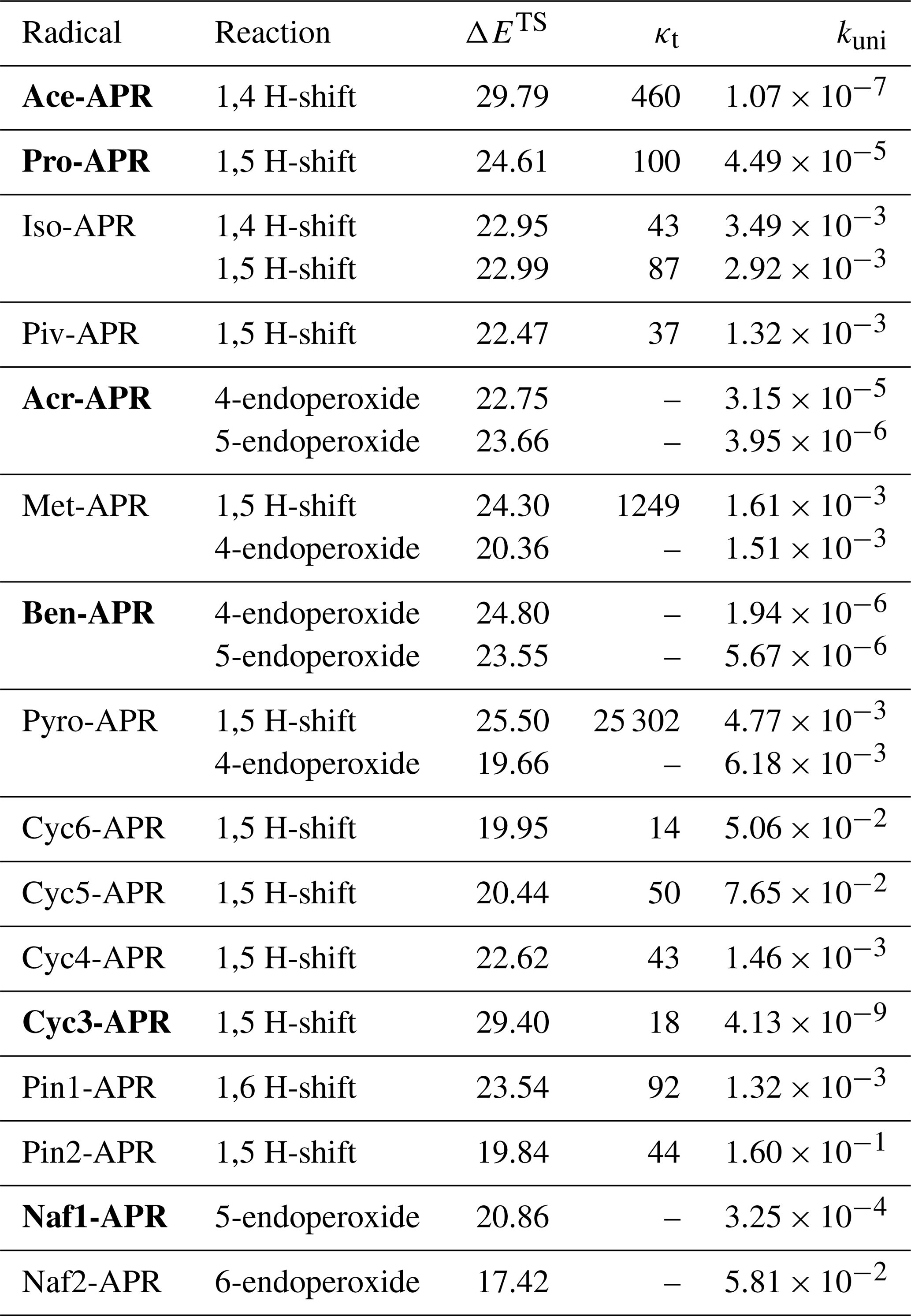
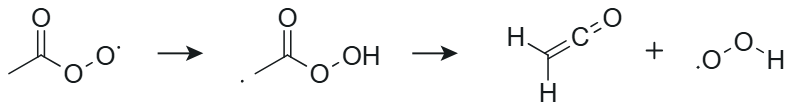
The slowest unimolecular reactions were observed for the smallest systems such as ace-APR and cyc3-APR. The optimization of the TS structure for the 1,4 H-shift reaction of ace-APR turned out to be difficult. A decomposition of the product results in the formation of ethenone and hydroperoxy radical during the optimization, making it difficult to find the correct TS. This decomposition channel of the product is presented in Fig. 3. The reaction was also studied in more detail by Sandhiya and Senthilkumar (2020). However, after several attempts, the correct TS structure was found and the correct rate coefficient was calculated for this H-shift reaction. For the pyro-APR 1,5 H-shift, a very large tunneling coefficient of 25 302 was observed. A high imaginary frequency of 2238i cm−1 results in a large tunneling coefficient. This coefficient differs significantly from other calculated tunneling coefficients, which could suggest that the Eckart method may no longer be reliable in this case. The Eckart method is a very simple model compared to multidimensional models that also take into account other variables along the reaction path (Zhang and Dibble, 2011; Meana-Pañeda et al., 2011). However, for the purposes of this study, using the Eckart method is reasonable due to the saved computational resources.
By increasing the system size, an increase in the unimolecular rate coefficients was observed, which was to be expected. A larger system size allows H-shifts from further positions relative to the peroxy radical group, resulting in a larger TS ring size with less strain. However, slower reactions were also observed for rigid systems, e.g., ben-APR and naf1-APR. This is due to the rigid structure and aromaticity of the benzene ring, which does not allow fast H-shift or endoperoxide ring formation reactions. However, naf2-APR is capable of quite fast unimolecular reactions despite the rigid structure. By adjusting the position of the APR group, a 6-endoperoxide ring formation reaction becomes possible, leading to the formation of a six-member ring product and facilitating a faster unimolecular reaction. A similar reaction mechanism is not efficient for naf1-APR, as the formation of the six-member ring is hindered.
Endoperoxide ring formations were found to be competitive with H-shift reactions or, in most cases, even faster. Vereecken et al. (2021) studied ring closure reactions and calculated rate coefficients for multiple different RO2 radicals, including C2H3CH2OO⋅, a non-acyl peroxy radical system similar to acr-APR. For C2H3CH2OO⋅, values of s−1 and s−1 were obtained for 4-endoperoxide and 5-endoperoxide reactions, respectively, in the study by Vereecken et al. (2021). The values obtained in this study for acr-APR are significantly higher compared to C2H3CH2OO⋅, suggesting that ring formation reactions are faster for APRs compared to other RO2. Unexpectedly, 4-endoperoxide reactions for acr-APR, met-APR, and pyro-APR were faster than the 5-endoperoxide reactions. In contrast, according to the values calculated by Vereecken et al. (2021) for C2H3CH2OO⋅, the 5-endoperoxide reaction was faster than the 4-endoperoxide reaction. The TS structures of acr-APR 4-endoperoxide and 5-endoperoxide reactions are presented in Fig. 4. Moreover, we did not observe any difference in allylic and non-allylic H-shift reactions when comparing the iso-APR and met-APR 1,5 H-shifts. For other RO2 radicals, allylic H-shifts have been shown to be significantly faster than non-allylic H-shifts (Otkjær et al., 2018; Vereecken and Nozière, 2020). However, we have been unable to identify any reasons for these distinct behaviors of APRs compared to other RO2 radicals.
3.2 Bimolecular reactions
Isoprene and peroxy radicals can react through four different pathways, where the peroxy radical adds to one of the four sp2 carbons in isoprene. These four pathways are presented in Fig. 5. Pasik et al. (2024b) studied these different pathways and showed that reaction pathways R1 and R4, where the peroxy radical adds to one of the two terminal sp2 carbons, are the fastest. Reaction pathway R1 leads to a tertiary (allylic) radical and reaction pathway R4 to a secondary (allylic) radical, which makes them faster than reaction pathways R2 and R3, which lead to primary radicals. Therefore, only reaction pathways R1 and R4 were the focus of this study.
Of interest were the APR structures, which had slow ( s−1) unimolecular reactions. Correspondingly, six APR structures (ace-APR, pro-APR, acr-APR, ben-APR, cyc3-APR, and naf1-APR) were chosen for further calculations to study their bimolecular reactions (see Table 1). The same reaction pathways were also studied for the reaction between isoprene and OH to assess the significance of APR-initiated oxidation of isoprene. Calculations of reactions between isoprene and APRs were carried out at the DLPNO-CCSD(T)/aug-cc-pVTZ//ωB97X-D/6-31+G* level, and calculations for OH reactions were carried out at the DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2X/6-31+G* level. Table 2 presents the calculated rate coefficients for reaction pathways R1 and R4. The table also includes the forward reaction barrier heights and the total reaction rates, which were assumed to be a sum of reaction rates R1 and R4.
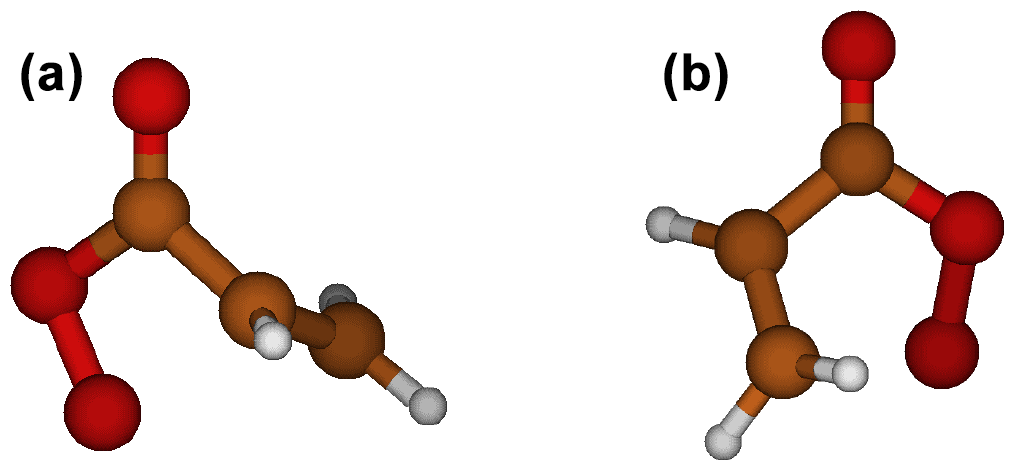
Figure 4Transition state structures of the acr-APR 4-endoperoxide (a) and 5-endoperoxide (b) ring formation reactions. Color coding: brown is carbon, red is oxygen, and white is hydrogen.
Table 2Calculated energy barrier heights (ΔETS; kcal mol−1) and bimolecular MC-TST reaction rate coefficients (kbi; cm3s−1) at 298 K for reactions R1 and R4 of isoprene with OH and APRs. Rates for APR and isoprene reactions were calculated at the DLPNO-CCSD(T)/aug-cc-pVTZ//ωB97X-D/6-31+G* level and, for OH and isoprene reactions, at the DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2X/6-31+G* level. The total kbi is the sum of reaction rates R1 and R4.
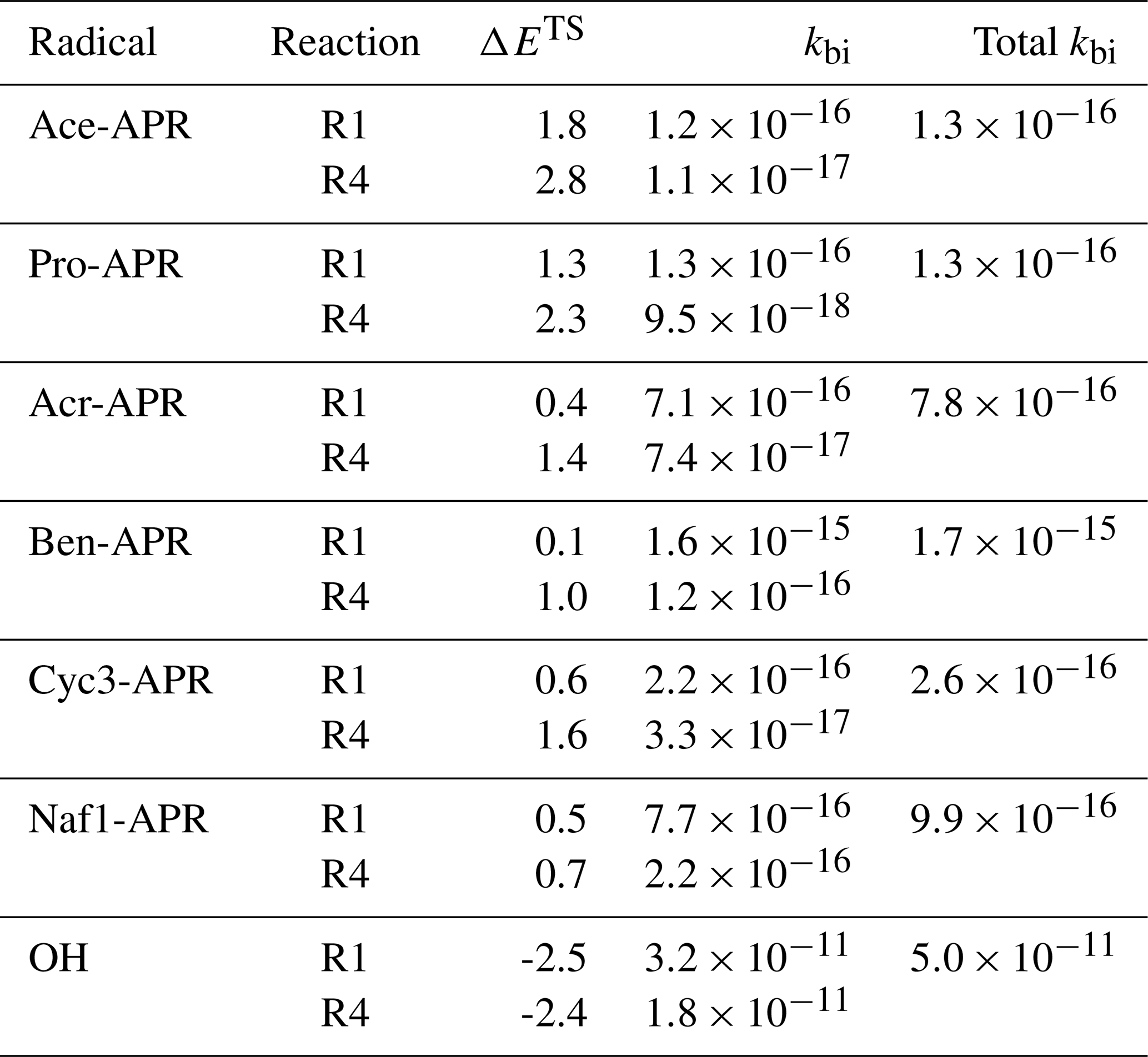
As expected, reaction pathway R1 is the fastest of all of the studied structures. This is explained by the reaction product having a tertiary resonance stabilized carbon-centered radical in comparison to reaction pathway R4, which leads to a secondary (allylic) radical. It can also be seen that the forward barrier decreases as the reactant APR size increases. Unsaturated APR, acr-APR, also exhibits faster reactions with isoprene. These results align with observations made in other studies for APRs and other RO2 radicals (Pasik et al., 2024b; Nozière and Fache, 2021). Unsaturated and large APRs could effectively stabilize the breaking of the π bond as the electron density can be delocalized over several atoms, resulting in decreased barriers. An exception to this is naf1-APR reaction R1, which has a slightly higher barrier than the corresponding reaction of ben-APR despite the larger structure of the naf1-APR reaction. However, the barrier for naf1-APR reaction R1 is still close to zero, resulting in a relatively fast reaction.
The values for the three reactions of isoprene with ace-APR, pro-APR, and ben-APR calculated at the DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2X/6-31+G* level are provided in Table S2. The rate coefficients calculated with the M06-2X functional are smaller, up to approximately an order of magnitude, compared to the ones calculated at the DLPNO-CCSD(T)/aug-cc-pVTZ//ωB97X-D/6-31+G* level. Therefore, the different levels of theory used in this study are not completely compatible with each other and make the comparison of OH- and APR-initiated isoprene oxidation more difficult. However, reasonable conclusions can be drawn from the results, and the impact of APR-initiated oxidation of isoprene can be assessed.
Nozière and Fache (2021) obtained an experimental value of cm3 s−1 for the reaction between ace-APR and isoprene. In the same study, a structure–activity–relationship (SAR) recommended by Stark (1997) was used to calculate a value of cm3 s−1 for the same reaction. Both rates are considerably higher than the total rate of cm3 s−1 calculated in this study. This would suggest that the rates calculated in this study underestimate the total bimolecular rates for reactions between APRs and isoprene. We also underestimate the rate constant for OH reaction with isoprene. An experimental value of cm3 s−1 (Atkinson et al., 1997) has been obtained for the reaction between OH and isoprene, which differs from our theoretical value by a factor of 2. The value calculated in this study does not take into account the other two reaction pathways R2 and R3, which also contribute to the rate coefficients. However, these contributions are minor and somewhere in the percentage range (Jenkin et al., 1998; Stevens et al., 2000). While we underestimate the rates for these reactions, the comparison between OH- and APR-initiated isoprene oxidation should be reliable.
The change in bimolecular reaction rates as a function of temperature was also investigated at the same levels of theory as the initial calculations. Rate coefficients were calculated at four different temperatures (248, 273, 298, and 323 K), and the total bimolecular rates were again assumed to be a sum of reaction rates R1 and R4. The pseudo-first-order reaction rates were calculated from the total bimolecular rates using a concentration of 106 cm−3 for OH (Wennberg et al., 2018) and a concentration of 108 cm−3 for all of the APRs. The APR concentration used is a mean value of the ace-APR concentration in hydrocarbon-rich remote atmospheres under low-NOx conditions from a study by Villenave et al. (1998). Data on other APR concentrations could not be obtained, and the ace-APR concentration was used as a default concentration for all of the other APRs. This assumption leads to some uncertainty in the results, but they can be assumed to be an upper limit for the pseudo-first-order rates. The results are provided in Table 3. An increase in the pseudo-first-order rates for the APR reactions can be observed as the temperature increases. By contrast, a decrease in the pseudo-first-order rate of reaction between isoprene and OH can be distinguished as the temperature increases. The negative temperature dependence of OH-initiated oxidation of isoprene has also been observed in experimental studies (Dillon et al., 2017; Kleindienst et al., 1982). This difference in the temperature dependence can be attributed to differences in the barriers. The addition of OH to isoprene has a negative barrier, whereas APR additions have positive barriers. This leads to a larger impact of APR-initiated oxidation of isoprene compared to that of OH at higher temperatures. At 248 K, APRs could be responsible for up to 0.1 % of isoprene oxidation compared to OH-initiated isoprene oxidation, and at 323 K the significance of APR-initiated oxidation could be up to 1 %.
Table 3Calculated pseudo-first-order reaction rates (kpseudo [radical]; cm−3 s−1) for studied reactions of isoprene with OH and APRs at four different temperatures (T; K). Concentrations of 106 molecules per cubic centimeter and 108 molecules per cubic centimeter were used for OH and all of the APRs, respectively.

We investigated unimolecular H-shift and endoperoxide ring formation reactions for a variety of acyl peroxy radicals using quantum-mechanical methods. The goal of this study was to determine which APRs could work as atmospheric oxidants of unsaturated hydrocarbons. We selected 16 APR structures with different functionalities and found 6 APRs with slow unimolecular reactions, making them potential oxidants of alkenes. As expected, many of the studied APRs had quite fast unimolecular reactions of up to 0.1 s−1. However, we found several APRs with low unimolecular reaction rate coefficients. For instance, small ace-APRs and pro-APRs have reaction rate constants on the order of 10−7 and 10−5 s−1, respectively. An even slower reaction was observed for a rigid APR with a three-member ring, cyc3-APR, for which the reaction rate constant was found to be as low as 10−9 s−1. For aromatic structures, the endoperoxide ring formation was faster than the H-shift reaction, but due to the rigid structure and aromaticity the reaction rate constants were still low, on the order of 10−5 s−1 for ben-APR and 10−4 s−1 for naf1-APR. Another naphthalene structure, naf2-APR, was able to form a six-member endoperoxide ring, and therefore the unimolecular endoperoxide ring formation reaction was fast. Generally, where endoperoxide ring formation reactions were possible, they were shown to be competitive with H-shift reactions and in most cases even faster.
Bimolecular accretion reactions between isoprene and APRs were also investigated in this study. Six APRs with slow unimolecular reactions were chosen for further calculations. Two pathways producing an allylic radical, R1 and R4, were studied for reactions between isoprene and the APRs. Pathway R1 was proven to be faster due to the formation of a tertiary allylic radical. Quite consistently, the reaction barrier heights decreased as the system size increased for both pathways. The temperature dependence of these reactions was also studied and compared to OH-initiated isoprene oxidation. We calculated the pseudo-first-order reaction rates for isoprene oxidation initiated by OH and six APRs at four different temperatures. For all of the studied APRs, the reaction rate increased as the temperature increased, whereas in the case of OH the reaction rate decreased as the temperature increased. This indicates a higher impact of APR-initiated isoprene oxidation compared to that of OH at higher temperatures. At 323 K, APRs could be responsible for up to 1 % of isoprene oxidation in the atmosphere compared to OH. While the percentage of APR-initiated oxidation of isoprene compared to OH is not significant, APR-initiated oxidation can lead directly to high-molar-mass dimeric products. These dimeric compounds with multiple oxygen atoms and high molar mass have low vapor pressure, and therefore they are candidates for participating in new particle formation and growth. Thus, despite the minor contribution to initiating oxidation of isoprene, APRs might play an important role in producing low-volatility organic compounds from reactions with other unsaturated hydrocarbons, such as monoterpenes. For a better understanding of the impact of APR-initiated oxidation, measurements contributing to APR concentrations in the atmosphere are needed.
The optimized structures and calculation output files of all the relevant compounds that support the findings of this paper will be available in the Zenodo repository: https://doi.org/10.5281/zenodo.15112993 (Karppinen, 2025).
The supplement related to this article is available online at https://doi.org/10.5194/ar-3-175-2025-supplement.
IK performed the calculations and wrote the manuscript. DP and EA assisted with the calculations. DP and NM contributed to the analysis. The study was designed and supervised by NM. All the authors proofread the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We acknowledge the CSC-IT Center for Science in Espoo, Finland, for the computational resources.
This research has been supported by the Research Council of Finland (grant no. 347775) and the Doctoral Programme in Atmospheric Sciences (ATM-DP), University of Helsinki.
This paper was edited by Jonas Elm and reviewed by three anonymous referees.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, 2003. a
Atkinson, R., Baulch, D., Cox, R., Hampson Jr., R., Kerr, J., Rossi, M., and Troe, J.: Evaluated kinetic, photochemical and heterogeneous data for atmospheric chemistry: Supplement V. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry, J. Phys. Chem. Ref. Data, 26, 521–1011, 1997. a, b
Bannwarth, C., Ehlert, S., and Grimme, S.: GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comput., 15, 1652–1671, 2019. a
Calvert, J., Mellouki, A., Orlando, J., Pilling, M., and Wallington, T.: Mechanisms of Atmospheric Oxidation of the Oxygenates, Oxford University Press USA, ISBN 9780199767076, 2011. a
Chai, J.-D. and Head-Gordon, M.: Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 10, 6615–6620, 2008a. a
Chai, J.-D. and Head-Gordon, M.: Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 128, 8, https://doi.org/10.1063/1.2834918, 2008b. a
Clark, T., Chandrasekhar, J., Spitznagel, G. W., and Schleyer, P. V. R.: Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+ G basis set for first-row elements, Li–F, J. Comput. Chem., 4, 294–301, 1983. a
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, 2013. a
Demore, W., Sander, S., Golden, D., Hampson, R., Kurylo, M., Howard, C., Ravishankara, A., Kolb, C., and Molina, M.: Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling, JPL Publication, 90, 196 pp., https://ntrs.nasa.gov/api/citations/19880014628/downloads/19880014628.pdf (last access: 11 November 2024), 1997. a
Dillon, T. J., Dulitz, K., Groß, C. B. M., and Crowley, J. N.: Temperature-dependent rate coefficients for the reactions of the hydroxyl radical with the atmospheric biogenics isoprene, alpha-pinene and delta-3-carene, Atmos. Chem. Phys., 17, 15137–15150, https://doi.org/10.5194/acp-17-15137-2017, 2017. a
Dunning Jr., T. H.: Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 90, 1007–1023, 1989. a
Eckart, C.: The penetration of a potential barrier by electrons, Phys. Rev., 35, 1303, https://doi.org/10.1103/PhysRev.35.1303, 1930. a
El-Agamey, A. and McGarvey, D. J.: Acyl/aroylperoxyl radicals: a comparative study of the reactivity of peroxyl radicals resulting from the α-cleavage of ketones, Phys. Chem. Chem. Phys., 4, 1611–1617, 2002. a
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery, Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., and Fox, D. J.: Gaussian 16 Revision C.02, gaussian Inc. Wallingford CT, 2016. a
Gu, C., Wang, S., Zhu, J., Wu, S., Duan, Y., Gao, S., and Zhou, B.: Investigation on the urban ambient isoprene and its oxidation processes, Atmos. Environ., 270, 118870, https://doi.org/10.1016/j.atmosenv.2021.118870, 2022. a
Hehre, W. J., Ditchfield, R., and Pople, J. A.: Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules, J. Chem. Phys., 56, 2257–2261, 1972. a
Jenkin, M. E., Boyd, A. A., and Lesclaux, R.: Peroxy radical kinetics resulting from the OH-initiated oxidation of 1, 3-butadiene, 2, 3-dimethyl-1, 3-butadiene and isoprene, J. Atmos. Chem., 29, 267–298, 1998. a
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019. a
Johnston, H. S. and Heicklen, J.: Tunnelling corrections for unsymmetrical Eckart potential energy barriers, J. Phys. Chem., 66, 532–533, 1962. a
Karppinen, I.: The impact of unimolecular reactions on acyl peroxy radical initiated isoprene oxidation, In Aerosol Research, Zenodo [data set], https://doi.org/10.5281/zenodo.15112994, 2025. a
Kendall, R. A., Dunning, T. H., and Harrison, R. J.: Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 96, 6796–6806, 1992. a
Kleindienst, T. E., Harris, G. W., and Pitts, J. N.: Rates and temperature dependences of the reaction of hydroxyl radical with isoprene, its oxidation products, and selected terpenes, Environ. Sci. Technol., 16, 844–846, 1982. a
Knap, H. C. and Jørgensen, S.: Rapid hydrogen shift reactions in acyl peroxy radicals, J. Phys. Chem. A, 121, 1470–1479, 2017. a, b, c
Kubečka, J., Besel, V., Kurtén, T., Myllys, N., and Vehkamaki, H.: Configurational sampling of noncovalent (atmospheric) molecular clusters: sulfuric acid and guanidine, J. Phys. Chem. A, 123, 6022–6033, 2019. a
Lee, S.-H., Gordon, H., Yu, H., Lehtipalo, K., Haley, R., Li, Y., and Zhang, R.: New particle formation in the atmosphere: From molecular clusters to global climate, J. Geophys. Res.-Atmos., 124, 7098–7146, 2019. a
McMahon, R. J.: Chemical reactions involving quantum tunneling, Science, 299, 833–834, 2003. a
Meana-Pañeda, R., Truhlar, D. G., and Fernández-Ramos, A.: High-level direct-dynamics variational transition state theory calculations including multidimensional tunneling of the thermal rate constants, branching ratios, and kinetic isotope effects of the hydrogen abstraction reactions from methanol by atomic hydrogen, J. Chem. Phys., 134, 9, https://doi.org/10.1063/1.3555763, 2011. a
Møller, K. H., Otkjær, R. V., Hyttinen, N., Kurtén, T., and Kjaergaard, H. G.: Cost-effective implementation of multiconformer transition state theory for peroxy radical hydrogen shift reactions, J. Phys. Chem. A, 120, 10072–10087, 2016. a, b
Møller, K. H., Bates, K. H., and Kjaergaard, H. G.: The importance of peroxy radical hydrogen-shift reactions in atmospheric isoprene oxidation, J. Phys. Chem. A, 123, 920–932, 2019. a, b
Møller, K. H., Otkjær, R. V., Chen, J., and Kjaergaard, H. G.: Double bonds are key to fast unimolecular reactivity in first-generation monoterpene hydroxy peroxy radicals, J. Phys. Chem. A, 124, 2885–2896, 2020. a
Neese, F.: The ORCA program system, Wiley Interdisciplinary Reviews: Computational Molecular Science, 2, 73–78, 2012. a
Nozière, B. and Fache, F.: Reactions of organic peroxy radicals, RO 2, with substituted and biogenic alkenes at room temperature: unsuspected sinks for some RO 2 in the atmosphere?, Chem. Sci., 12, 11676–11683, 2021. a, b, c, d
Nozière, B. and Vereecken, L.: H-shift and cyclization reactions in unsaturated alkylperoxy radicals near room temperature: propagating or terminating autoxidation?, Phys. Chem. Chem. Phys., 26, 25373–25384, 2024. a
Nozière, B., Durif, O., Dubus, E., Kylington, S., Emmer, Å., Fache, F., Piel, F., and Wisthaler, A.: The reaction of organic peroxy radicals with unsaturated compounds controlled by a non-epoxide pathway under atmospheric conditions, Phys. Chem. Chem. Phys., 25, 7772–7782, 2023. a
Otkjær, R. V., Jakobsen, H. H., Tram, C. M., and Kjaergaard, H. G.: Calculated hydrogen shift rate constants in substituted alkyl peroxy radicals, J. Phys. Chem. A, 122, 8665–8673, 2018. a
Pasik, D., Frandsen, B. N., Meder, M., Iyer, S., Kurtén, T., and Myllys, N.: Gas-Phase Oxidation of Atmospherically Relevant Unsaturated Hydrocarbons by Acyl Peroxy Radicals, J. Am. Chem. Soc., 146, 13427–13437, 2024a. a
Pasik, D., Iyer, S., and Myllys, N.: Cost-effective approach for atmospheric accretion reactions: a case of peroxy radical addition to isoprene, Phys. Chem. Chem. Phys., 26, 2560–2567, 2024b. a, b, c
Pracht, P., Bohle, F., and Grimme, S.: Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 22, 7169–7192, 2020. a
Riplinger, C. and Neese, F.: An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys., 138, 3, https://doi.org/10.1063/1.4773581, 2013. a
Riplinger, C., Sandhoefer, B., Hansen, A., and Neese, F.: Natural triple excitations in local coupled cluster calculations with pair natural orbitals, J. Chem. Phys., 139, 13, https://doi.org/10.1063/1.4821834, 2013. a
Sandhiya, L. and Senthilkumar, K.: Unimolecular decomposition of acetyl peroxy radical: a potential source of tropospheric ketene, Phys. Chem. Chem. Phys., 22, 26819–26827, 2020. a
Seal, P., Barua, S., Iyer, S., Kumar, A., and Rissanen, M.: A systematic study on the kinetics of H-shift reactions in pristine acyl peroxy radicals, Phys. Chem. Chem. Phys., 25, 28205–28212, 2023. a, b
Stark, M. S.: Epoxidation of alkenes by peroxyl radicals in the gas phase: structure- activity relationships, J. Phys. Chem. A, 101, 8296–8301, 1997. a, b
Stevens, P. S., Seymour, E., and Li, Z.: Theoretical and experimental studies of the reaction of OH with isoprene, J. Phys. Chem. A, 104, 5989–5997, 2000. a
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020. a, b, c
Vereecken, L. and Peeters, J.: The 1, 5-H-shift in 1-butoxy: A case study in the rigorous implementation of transition state theory for a multirotamer system, J. Chem. Phys., 119, 5159–5170, 2003. a
Vereecken, L. and Peeters, J.: Nontraditional (per) oxy ring-closure paths in the atmospheric oxidation of isoprene and monoterpenes, J. Phys. Chem. A, 108, 5197–5204, 2004. a
Vereecken, L., Vu, G., Wahner, A., Kiendler-Scharr, A., and Nguyen, H.: A structure activity relationship for ring closure reactions in unsaturated alkylperoxy radicals, Phys. Chem. Chem. Phys., 23, 16564–16576, 2021. a, b, c, d, e
Viegas, L. P.: Exploring the reactivity of hydrofluoropolyethers toward OH through a cost-effective protocol for calculating multiconformer transition state theory rate constants, J. Phys. Chem. A, 122, 9721–9732, 2018. a
Viegas, L. P.: Simplified protocol for the calculation of multiconformer transition state theory rate constants applied to tropospheric OH-initiated oxidation reactions, J. Phys. Chem. A, 125, 4499–4512, 2021. a
Villenave, E., Lesclaux, R., Seefeld, S., and Stockwell, W. R.: Kinetics and atmospheric implications of peroxy radical cross reactions involving the CH3C (O) O2 radical, J. Geophys. Res.-Atmos., 103, 25273–25285, 1998. a
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, 2018. a
Xu, L., Møller, K. H., Crounse, J. D., Otkjær, R. V., Kjaergaard, H. G., and Wennberg, P. O.: Unimolecular reactions of peroxy radicals formed in the oxidation of α-pinene and β-pinene by hydroxyl radicals, J. Phys. Chem. A, 123, 1661–1674, 2019. a
Zhang, F. and Dibble, T. S.: Impact of tunneling on hydrogen-migration of the n-propylperoxy radical, Phys. Chem. Chem. Phys., 13, 17969–17977, 2011. a
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Account., 120, 215–241, 2008. a