the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 15 Apr 2025
| 15 Apr 2025
Investigation of soot precursor molecules during inception by acetylene pyrolysis using reactive molecular dynamics
Anindya Ganguly
Khaled Mosharraf Mukut
Somesh Roy
Georgios Kelesidis
Eirini Goudeli
Soot inception by acetylene pyrolysis at 1350–1800 K is investigated using reactive molecular dynamics. The composition and chemical structure of soot precursor molecules formed during inception are elucidated. During soot inception, increasing the process temperature leads to faster depletion of C2H2 molecules and faster formation of C2H3, C2H4, C2H6, CH4, and C2 with the concurrent appearance of H2 molecules. Small molecules consisting of 1–5 C atoms (C1–C5) are formed due to reactive collisions and grow further to larger hydrocarbon compounds consisting of 6–10 C atoms. At initial stages of inception, prior to the formation of incipient soot, three-member rings are formed, which are associated with the formation of compounds with fewer than 10 C atoms. Once incipient soot is formed, the number of C1–C10 compounds and the number of three-member rings drop, while the number of five- and six-member rings increases, indicating that the formation of larger rings is associated with the growth of soot clusters. The chemical structure of soot precursor molecules obtained by bond order analysis reveals that molecules with up to 10 C atoms are either linear or branched aliphatic compounds or may contain three-member rings fused with aliphatic components. Molecules with more than 10 C atoms often exhibit structures composed of five- or six-member C rings, decorated by aliphatic components. The identification of molecular precursors contributing to soot inception provides crucial insights into soot formation mechanisms, pinpointing potential pathways of soot formation during combustion.
- Article
(5756 KB) - Full-text XML
-
Supplement
(5944 KB) - BibTeX
- EndNote




Soot is formed during incomplete combustion or pyrolysis of hydrocarbons (Michelsen et al., 2020) and exhibits adverse effects on human health (Anenberg et al., 2012), leading to respiratory diseases (Shiraiwa et al., 2017) and premature deaths (Giannadaki et al., 2016). Soot is a large contributor to global warming after CO2 (Bond et al., 2013), affecting local, regional, and global climate (Ramanathan et al., 2001). Formation of soot is believed to take place by gas-phase condensation or reaction of precursor molecules and polycyclic aromatic hydrocarbons (PAHs) (Johansson et al., 2018), leading to incipient soot (inception) (Wang and Chung, 2019), which grows further by condensation of gas-phase radicals on its surface (surface growth) (Michelsen et al., 2020). These primary soot nanoparticles collide with each other, forming aggregates that can break upon oxidation (Naseri et al., 2022). Even though soot growth by coagulation is rather well understood through advances in numerical modeling (Kazakov and Frenklach, 1998; Maricq, 2007; Sun et al., 2021), simulations (Kelesidis et al., 2017a, b; Kelesidis and Goudeli, 2021), and measurements (Maricq, 2007; Rissler et al., 2013; Maricq, 2014), development of an accurate physical representation of the early stages of soot formation, and particularly inception (Irimiea et al., 2019), possess significant challenges to kinetic modeling of soot (Appel et al., 2000), as it requires knowledge of the chemical reaction pathways from gaseous species to soot clusters (Thomson, 2023). Thus, a more detailed molecular-level understanding of the species formed during soot inception is essential for the accurate description of those chemical reaction pathways and for the design of selective, soot-free chemical processes.
Kinetic models (Frenklach and Wang, 1991, 1994) often assume that soot inception occurs through physical PAH dimerization. This is in contrast to theoretical calculations (Houston Miller et al., 1985; Miller, 1991; Schuetz and Frenklach, 2002), revealing much lower PAH concentrations than those of small soot particles (Houston Miller et al., 1985) with short lifetimes (< 75 ps) for PAH dimers smaller than 800 amu (Schuetz and Frenklach, 2002; Miller, 1991). Such theoretical calculations are consistent with measurements (Sabbah et al., 2010) of the kinetics of pyrene dimerization, corroborating the finding that physical PAH dimerization does not contribute significantly to soot inception. Recent research on combustion has been focusing on chemical dimerization of PAHs through the formation of covalent bonds (Johansson et al., 2017), with experiments suggesting that acetylene (C2H2) or vinyl (C2H3) addition via radical chain reaction leads to molecular growth of PAHs via chemisorption (Johansson et al., 2018). Additionally, shock tube experiments of C2H2 and C4H2 pyrolysis (Kiefer et al., 1992) and ab initio simulations of C2H2 (Zádor et al., 2017) have shown that pyrolysis of aliphatic hydrocarbons produces C2Hx radicals/intermediates, such as acetylene/ethynyl (C2H2/C2H) and ethylene/vinyl (C2H4/C2H3) (Tanzawa and Gardiner, 1980) at high concentrations, which either initiate or accelerate the formation of a wide variety of products including cyclopentaring-fused PAHs (Shukla and Koshi, 2012). Density functional theory calculations have shown that acetylene–acetylene reaction via vinylidene formation leads to methylene cyclopropene, a three-member ring structure that is rapidly converted into aliphatic isomers (Zádor et al., 2017), indicating a resonance-stabilized hydrocarbon radical chain reaction pathway (Johansson et al., 2018). These findings suggest that reactions of acetylene are involved in nearly all hydrocarbon fuel pyrolysis processes (Liu et al., 2021) and pure acetylene pyrolysis serves as the basis for understanding pyrolysis of other hydrocarbons to soot formation (Slavinskaya et al., 2019).
Reactive molecular dynamics (MD) simulations based on the ReaxFF (reactive force field; Castro-Marcano et al., 2012) provide insight into the reaction kinetics (Schmalz et al., 2024) and dynamic formation of soot (Goudeli, 2019). Schmalz et al. (2024) identified the reaction pathways to the formation of benzene, predicted by ReaxFF pyrolysis of n-heptane and iso-octane, revealing that > 90 % of ReaxFF-obtained reactions were not considered in kinetic models (Langer et al., 2023). Such reaction path identification analysis, however, has only been limited to short simulation times and 100 fuel molecules, as even such small pyrolysis systems yield a complex network of more than 10 000 reactions. ReaxFF simulations have been used more commonly to investigate soot inception (Mao et al., 2017; Yuan et al., 2019; Han et al., 2017) and growth (Yuan et al., 2019) using PAHs as the starting nucleating species. These simulations (Mao et al., 2017) revealed that above 2000 K, PAHs grow into soot particles via chemical reactions, while below the boiling/sublimation temperature of the nucleating PAHs, soot inception occurs via physical PAH dimerization. The presence of large PAHs (> 398 amu) facilitates physical inception of smaller PAHs (< 202 amu) at low temperature (1000 K), but such PAHs dissociate at high temperature (> 1600 K), where soot clusters are formed by radical–radical reactions (Yuan et al., 2019). Arvelos et al. (2019) further demonstrated that at ∼ 2165 K, cyclohexanone undergoes molecular decomposition, leading to ethene and ethenone formation, marking a shift from stable product formation to radical-dominated process. Above 1500 K, addition of C2H2 leads to formation of bridges among PAHs and other unsaturated aliphatic hydrocarbons that compose the soot clusters (Yuan et al., 2019). Liu et al. (2020) expanded on this by investigating n-decane pyrolysis by ReaxFF simulations above 2300 K, showing that soot growth accelerates especially above 3000 K, where complete soot particle development and graphitization occur.
At 3000 K, reactive collisions of a multicomponent fuel revealed the formation of aliphatic polyyne-like chains that cyclized to form large rings, while internal bridging between macrocyclic carbon atoms led to PAHs with aliphatic side chains (Han et al., 2017). Also, PAH-like molecules coalesce via a ring-closing mechanism, leading to conversion of five- or seven-member rings to six-member ones (graphitization) with three-member rings as intermediates (Han et al., 2017). Similarly, Zhang et al. (2023) found that at 3000 K, 2,5-dimethylfuran (DMF) pyrolysis promotes rapid polycyclic aromatic hydrocarbon (PAH) growth, driven by increased dehydrogenation and active site availability. Sharma et al. (2021) observed that at low temperatures (< 1200 K), soot nanoparticles obtained by C2H2 pyrolysis are more prone to coalescence than at higher temperature, consistent with PAH-based clusters (Hou et al., 2022) due to low aromatic-to-aliphatic and ratios. Most of these reactive MD studies of soot or carbon black formation, however, are limited to high-temperature regions (> 1800 K) and usually consider common PAH molecules as precursors to soot formation (Zhao et al., 2020). So, the detailed molecular structures of soot precursors contributing to the chemical pathway to soot formation have not been thoroughly investigated, particularly at lower temperatures more relevant to combustion conditions.
Here, inception and growth of incipient soot are studied during pyrolysis using acetylene as initial fuel. This study investigates the detailed chemical structure of a wide range of soot precursors, providing a comprehensive analysis by accounting for detailed bond information obtained from ReaxFF MD simulations during acetylene pyrolysis at 1350–1800 K. The temporal evolution of the concentration of the most abundant species formed during inception is elucidated along with the average ratio of small (composed of fewer than 5 C atoms), intermediate (6–10 C atoms), and large molecules (> 10 C atoms) until the formation of super critical nuclei with up to 70 C atoms. The detailed chemical structure of the soot precursors at different stages of soot inception and surface growth is quantified based on bond order analysis, and the effect of temperature on the concentration and structure of these precursors is investigated.
2.1 Inception simulations
A total of 1000 acetylene (C2H2) molecules are randomly distributed in a cubic simulation cell of 75.6 Å length with periodic boundary conditions, using MAPS 4.3. The use of 1000 C2H2 molecules ensures sufficient statistical sampling of the population of precursors formed during pyrolysis. The simulation box size was chosen such that 1000 acetylene molecules achieve a density of 0.1 g cm−3 within the simulation domain. This density is consistent with that employed in other ReaxFF studies (Sharma et al., 2021), ensuring that incipient soot formation can occur within the timescale accessible by reactive MD simulations for the temperature range used in this study. Here, incipient soot refers to soot clusters with Mw ≥ 202 g mol−1 (Mukut et al., 2023), corresponding to the molecular weight of pyrene (Dillstrom and Violi, 2017), which is one of the most commonly considered seed molecules that initiate soot inception (Frenklach, 2002; Mukut et al., 2023). Bond breakage and new bond formation upon molecular collisions are simulated by employing the reactive force field of Castro-Marcano et al. (2012) for hydrocarbons, which has demonstrated consistency with experimental observations (Rokstad et al., 2014; Agafonov et al., 2015; Aghsaee et al., 2014) and theoretical models (Gao and Tang, 2022; Saggese et al., 2014; Slavinskaya et al., 2019; Liu et al., 2021), particularly in understanding radical chain mechanisms in acetylene pyrolysis. The bond lengths of the molecules are constantly adjusted based on their changing local chemical environment (Chenoweth et al., 2008). ReaxFF enables the simulation of chemically reactive systems through the prediction of atomic connectivity through interatomic distances, angles, and torsion terms. The total system energy is divided into various partial contributions, including bond energy (Ebond), over-coordination energy penalty (Eover), under-coordination stability (Eunder), valence angle energy (Eval), lone pair energy (Elp), penalty energy term (Epen), torsion angle energy (Eta), conjugation effects to molecular energy (Econj), van der Waals energy (EvdW), and Coulomb energy (ECoul):
The equations of motion are integrated using a velocity Verlet algorithm (Swope et al., 1982) with a time step of 0.25 fs (Chenoweth et al., 2009), consistent with ReaxFF MD simulations (Mao et al., 2017; Lümmen, 2010; Rom et al., 2013) of hydrocarbon reactions for soot formation at low temperature, using the Nosé–Hoover thermostat (Evans and Holian, 1985), with a damping parameter of 10 fs. Acetylene pyrolysis simulations are carried out for 10 ns in the NVT (constant number, volume, and temperature) ensemble using LAMMPS (Plimpton, 1995) at 1350, 1500, 1650, and 1800 K. The employed temperature range is consistent with temperature measurements during pyrolysis of ethylene (Dewa et al., 2016; Mei et al., 2019) and acetylene (Drakon et al., 2021). The simulation results were reproduced with up to three additional NVT inception simulations with different initial C2H2 configurations at each temperature (see the Supplement). The ReaxFF MD simulation details are listed in Table 1.

2.2 Chemical structure of soot precursor molecules
The chemical structure of all species consisting of up to 70 C atoms formed during pyrolysis and their number concentration are obtained as a function of time and are recorded every 0.25 ns. The detailed structure of each individual molecule is visualized using the Chemical Trajectory Analyzer (ChemTraYzer) analysis tool (Döntgen et al., 2015, 2018), which utilizes the bond order information and the atom coordinates generated by ReaxFF simulations. The individual molecules emerging at each time step are distinguished based on the atom connectivity data available in the bond order files generated by ReaxFF simulations, and their detailed chemical structure is visualized based on the ReaxFF-obtained bond order information based on their interatomic distances (Chenoweth et al., 2008). In ChemTraYzer, the bond orders are rounded to increments of 0.5 (i.e., 0.5, 1, 1.5, etc.) with those below 0.5 being disregarded, as discussed in Krep et al. (2022). The bond information is converted to simplified molecular-input line-entry system (SMILES) codes through Open Babel (O'Boyle et al., 2011) representing the detailed chemical structures of each of the molecules, which are visualized with MolView (Bergwerf, 2015). The snapshots of the entire simulated system were visualized using visual MD (VMD; Humphrey et al., 1996).
2.3 Analysis of cyclic structures
The total number of three-, five-, and six-member rings formed in the system is quantified at different time steps during the pyrolysis simulations. A connectivity matrix is constructed utilizing the coordinates and bond order of all atoms and their neighbors. When 3, 5, or 6 atoms are connected in series in a closed loop, they correspond to three-, five-, and six-member rings, respectively, and are distinguished from non-cyclic structures.
Once the cyclic structures have been identified, the bonds of each of their constituent pairs are categorized as single C–C bonds if the bond order is < 1.33 (Emri and Lente, 2004); double C–C bonds if the bond order values range between 1.33 and 2.85 (Emri and Lente, 2004; Hermann and Frenking, 2016); and triple C–C bonds if the bond order is at least 2.86, corresponding to the triple C–C bond in C2H2 (Hermann and Frenking, 2016). A bond order greater than or equal to 1.33 (graphite) (Emri and Lente, 2004) indicates the presence of a double bond for aromatic compounds, and a bond order of ∼ 1.92 (ethylene) indicates double bonds in aliphatic compounds (Hermann and Frenking, 2016). The aromaticity of each of the ring structures is assessed by counting the electrons in p orbitals that are involved in double bonds or lone pairs and applying Hückel's rule (Solà, 2022). Due to shared electron distribution and fractional bonds predicted by ReaxFF, along with the assignment of bond orders in increments of 0.5 by ChemTraYzer, some molecules might be visualized as having C atoms with more than four bonds, which have been excluded from the reported results. The four- and seven-member ring structures are determined by MAFIA-MD (molecular arrangement and fringe identification and analysis from molecular dynamics; Mukut et al., 2022).
3.1 Molecular properties of soot precursors
Figure 1 shows snapshots of the growth of carbonaceous nanoparticles formed by acetylene pyrolysis at (a) 1350, (b) 1500, (c) 1650, and (d) 1800 K at times t = 0.75, 2, 3.75, 5, 6, and 8 ns. All C–C bonds are represented by black lines, and all H atoms are omitted for clarity. Initially (t = 0.75 ns), at low temperatures (1350 and 1500 K), acetylene molecules hardly react with each other. At 1350 K, a few linear molecules appear at 2 and 3.75 ns (Fig. 1a: red-circled molecules) due to the reactive collisions of acetylene molecules, which grow into cyclic hydrocarbons at 5 ns (Fig. 1a: green-circled molecules) and later (t = 6 ns) into the incipient soot (Fig. 1a: blue-circled cluster). At 1500 K, linearization is observed at 2 ns (Fig. 1b: red-circled molecules), while cyclization takes place earlier than 1350 K, at 3.75 ns (Fig. 1b: green-circled clusters). After cyclization, the chain and cyclic molecules grow rapidly, forming a large incipient soot nanoparticle at 6 and 5 ns for 1350 and 1500 K, respectively, by scavenging the surrounding reactive molecules around the cluster (Fig. 1a and b: blue-circled clusters). These clusters grow further by surface growth (8 ns for both 1350 and 1500 K) until most of the surrounding reactive molecules are depleted.
At high temperature (T = 1650 and 1800 K), acetylene molecules collide more vigorously with each other due to their higher kinetic energy, increasing the probability of bond breakage upon collision. So, linearization and cyclization (Fig. 1c and d: red- and green-circled molecules) occur faster (t = 0.75 ns) at 1650 and 1800 K than at lower temperatures (Fig. 1a and b). Incipient soot forms rapidly within 2 ns (Fig. 1c and d: blue-circled molecules at t = 2 ns) and grows further by surface condensation for t ≥ 3.75 ns until the surrounding molecules and radicals are consumed. Therefore, higher temperature leads to faster hydrocarbon cyclization and growth, consistent with diffused back-illumination extinction imaging measurements of pyrolytic decomposition of n-dodecane (Skeen and Yasutomi, 2018), revealing a linear increase in the soot formation rate with temperature.
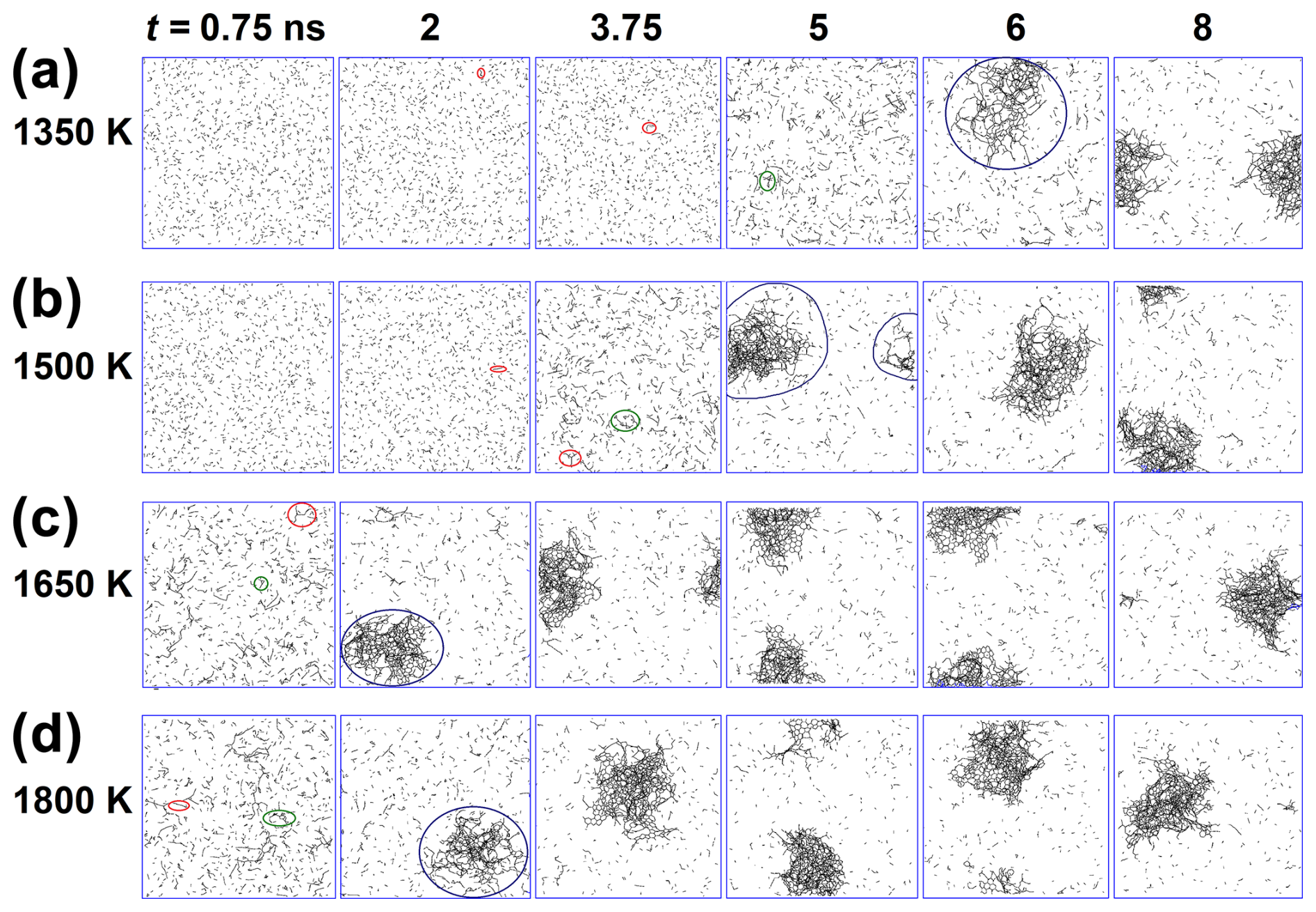
Figure 1Snapshots of the growth of carbonaceous species (hydrogen atoms are omitted) formed by acetylene pyrolysis at (a) 1350, (b) 1500, (c) 1650, and (d) 1800 K at times t = 0.75, 2, 3.75, 5, 6, and 8 ns. Formation of linear-like (chain) molecules (red-circled molecules) is observed early on, especially at high temperatures, followed by the formation of cyclic hydrocarbons (green-circled molecules). These linear and cyclic molecules coalesce to form incipient soot (blue-circled clusters), which grows further by the surface condensation of small free molecules or radicals.
Figure 2 shows the temporal evolution of the molecular weight of the largest molecule or cluster formed during acetylene pyrolysis at 1350 (black line), 1500 (blue line), 1650 (green line), and 1800 K (red line) for the simulations shown in Fig. 1. Initially, only acetylene molecules are present in the simulation domain corresponding to 26 g mol−1 at t = 0 ns at all temperatures. During acetylene pyrolysis, inception takes place slowly by reactive collisions of hydrocarbon molecules. The formation of incipient soot, defined as soot clusters with molecular weight equal to or greater than that of pyrene (Dillstrom and Violi, 2017), is denoted by the horizontal line in Fig. 2, corresponding to Mw = 202 g mol−1. The onset of surface growth as a function of temperature is shown in Fig. S1 in the Supplement. In addition, upon the formation of a molecule with molecular weight of ∼ 202 g mol−1, no dissociation of this molecule is observed (as shown exemplarily for T = 1800 K, Fig. S2), indicating the transition from gas to particle phase and the onset of surface growth. This lack of dissociation for clusters larger than ∼ 202 g mol−1 is observed for all simulations and temperatures. The inception step is slower at low temperature due to low kinetic energy of the colliding reactive species. For example, at 1350 K inception is completed within ∼ 5 ns, i.e., 5 times slower than inception at T ≥ 1650 K, which takes place within ∼ 1 ns.
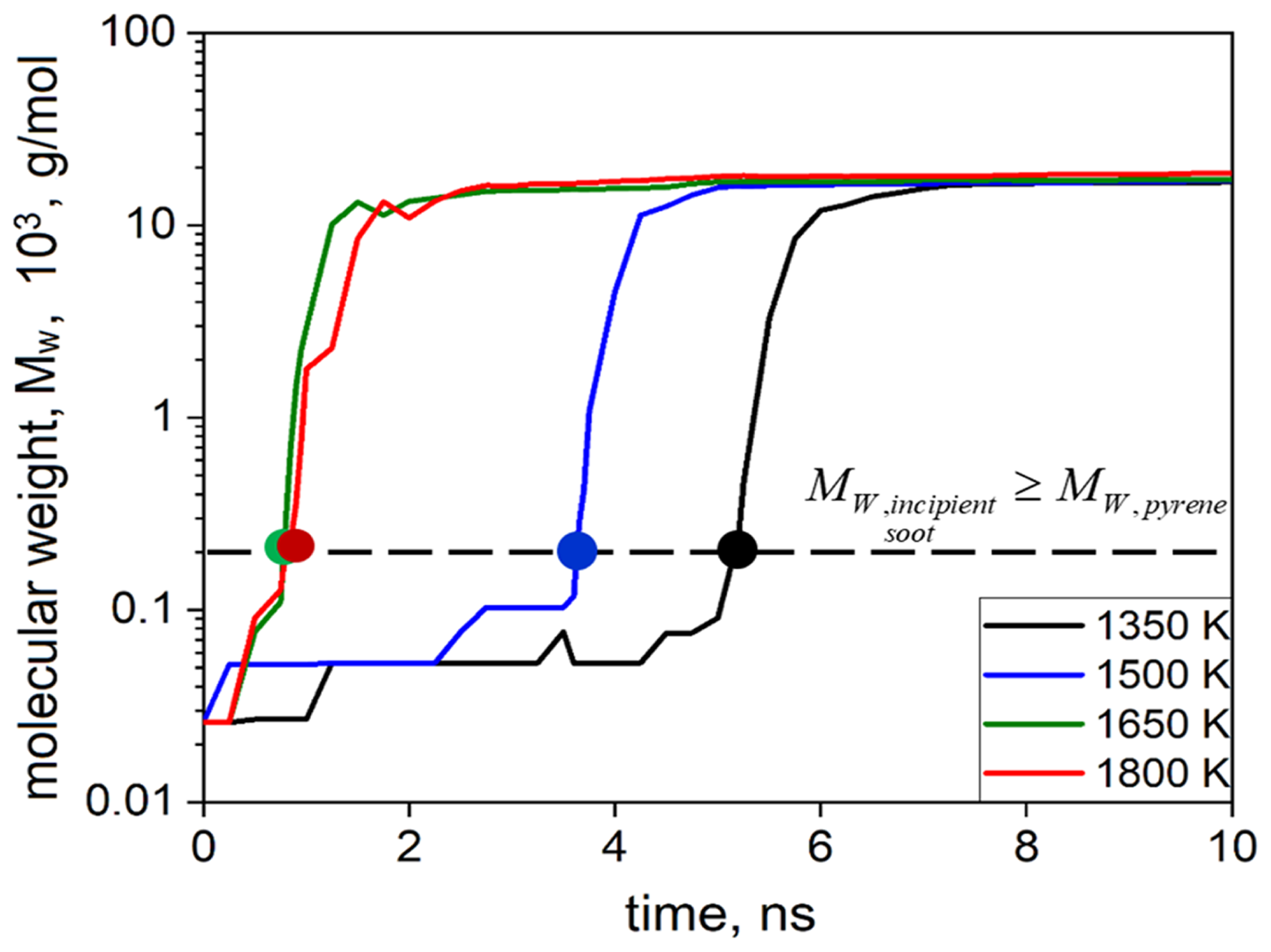
Figure 2Temporal evolution of the molecular weight of the largest molecule formed during acetylene pyrolysis at 1350 (black line), 1500 (blue line), 1650 (green line), and 1800 K (red line) for the simulations of Fig. 1. The formation of incipient soot is denoted by the formation of soot clusters with Mw ≥ 202 g mol−1 (horizontal line), corresponding to the molecular weight of the pyrene molecule.
Figure 3 shows the temporal evolution of the total number of (a) C2H2 molecules, as well as compounds containing (b) 1–5 C atoms (C1–C5; excluding C2H2), (c) 6–10 C atoms (C6–C10), and (d) > 10 C atoms (C10) present in the simulation cell, during C2H2 pyrolysis at 1350 (circles), 1500 (squares), 1650 (diamonds), and 1800 K (stars). The total number of C1–C5 molecules (including C2H2) is shown in Fig. S3. At T = 1350 K, the consumption of C2H2 is negligible (< 10 %) up to 4.75 ns. Shortly thereafter, at t = 5.2 ns (Fig. 3a: vertical black line), a sudden drop of 92.2 % in the C2H2 concentration is observed, accompanied by an abrupt increase in the number of other C1–C5 (Fig. 3b: circles) and C6–C10 molecules (Fig. 3c: circles). At that time (t = 5.2 ns), a C19H11 cluster is formed (Table S1, T = 1350 K: simulation 1), with a molecular weight of 239 g mol−1 denoting the onset of surface growth (vertical black line), as discussed in Fig. 2.
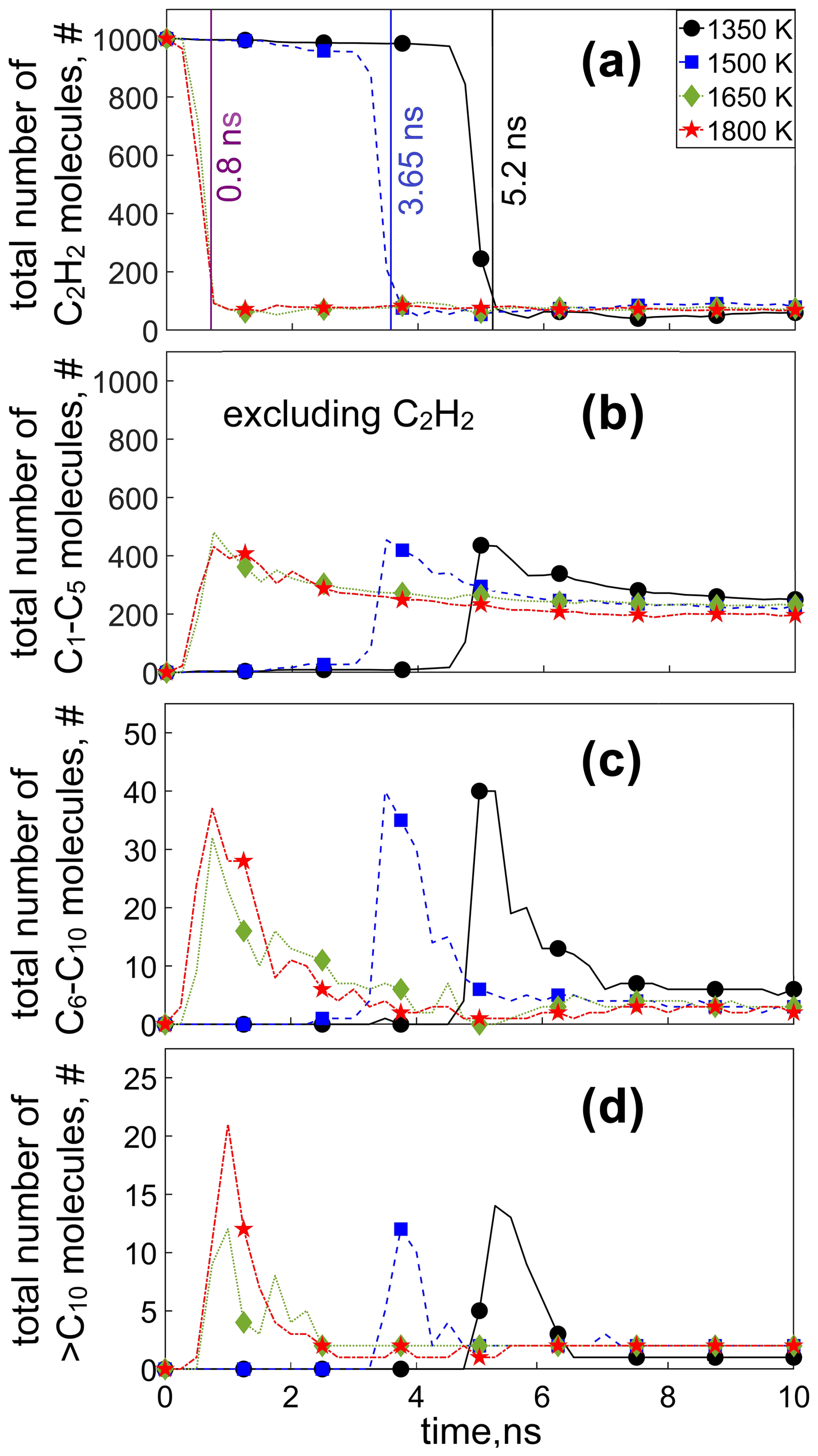
Figure 3Temporal evolution of the total number of (a) acetylene (C2H2), (b) C1–C5 (excluding C2H2), (c) C6–C10, and (d) > C10 molecules during acetylene pyrolysis at 1350 (circles), 1500 (squares), 1650 (diamonds), and 1800 K (stars). The vertical lines at 0.8 ns for 1650 and 1800 K, at 3.65 ns for 1500 K, and at 5.2 ns for 1350 K indicate the onset of surface growth for the respective temperatures.
The peak in the number of C6–C10 molecules (Fig. 3c) coincides with the increase in the number of C1–C5 molecules other than C2H2 (Fig. 3b) and the depletion of nearly 90 % of the C2H2 molecules (Fig. 3a). In fact, narrow peaks appear in the evolution of the number of C6–C10, indicating their rapid, practically complete consumption shortly after their formation. These intermediate-sized molecules (C6–C10) contribute up to 2.5 % of all molecules at the surface growth stage. In contrast, C1–C5 molecules excluding acetylene (Fig. 3b) are consumed at a slow rate, reaching a plateau of about 250 molecules at longer times (t > 8 ns), totaling 70 %–75 % of all molecules, regardless of the temperature. The faster consumption rate of the C6–C10 molecules could be attributed to their larger projected area compared to C1–C5 compounds, rendering them more likely to be scavenged by the large incipient soot. For example, the collision frequency function of a naphthalene molecule is approximately 1.6 times smaller than that of an acetylene molecule, based on the kinetic theory of gases. It should be noted, however, that the reactivity and chemical stability of each species vary depending on their size and chemical nature, which also affects their consumption rate.
At the onset of surface growth (t = 5.2 ns at 1350 K, Fig. 3a: vertical black line), the number concentration of larger molecules, composed of more than 10 C atoms (Fig. 3d: circles), also increases, reaching a maximum at t = 5.25 ns. As the largest cluster in the simulation domain (e.g., Fig. 1a: t = 6 ns) collides with the surrounding species, it grows by scavenging other molecules with a large projected area, resulting in a drop in the number of C6–C10 (Fig. 3c) and > C10 molecules. In contrast to larger molecules consisting of more than 6 C atoms, the concentration of C1–C5 molecules (Fig. 3b: circles) decreases at a much slower rate as the reactive collisions of such molecules with each other and with the soot cluster become more scarce. In fact, for t ≥ 6.5 ns, only one cluster with > C10 is present (Fig. 3d), corresponding to the incipient soot particle (composed of 1134 C atoms).
At higher temperature (T > 1350 K), even though soot precursor species form earlier than at 1350 K, a similar growth mechanism is observed. Specifically at T = 1500 K, the onset of surface growth takes place at 3.65 ns, corresponding to acetylene reduction of 87.6 % (Fig. 3a: vertical blue line) and to a peak in the number of C1–C5 (Fig. 3b) and C6–C10 molecules (Fig. 3c). This delay in the peak concentration of the C1–C10 molecules at low temperature is associated with the slower reaction kinetics of acetylene molecules, as indicated by their delayed depletion (Fig. 3a) compared to higher temperature. For t < 3.65 ns, small (< C10) molecules are mostly formed and only few (fewer than 10; Fig. 3d) larger molecules are present. At even higher temperature (T ≥ 1650 K), inception takes place at a much shorter timescale as the onset of surface growth is observed at t = 0.8 ns (Fig. 3a: vertical purple line), with a simultaneous increase in C1−10 molecules (Fig. 3b and c), which grow further after 0.2 ns, indicated by the peak of the > C10 molecules (Fig. 3d) at t = 1 ns.
At sufficiently long times, soot inception and surface condensation are practically completed as the number concentration of all species in the simulation domain is significantly reduced, reaching steady state. At this stage, 16 %–20 % of all the molecules are C2H2, 77.5 %–81.5 % are C1–C5 (excluding C2H2), and 2.5 % are C6–C10 molecules. These MD results have been reproduced with up to four simulations at each temperature using different initial configurations of acetylene molecules (Figs. S4–S9, simulations S1–S4), confirming the trends shown in Fig. 3. At 1350 K, a large variation is observed in the C2H2 evolution. The C2H2 molecules are practically consumed within 3.5 to 8 ns, following a similar trend in all four simulations (Fig. S4) and resulting in qualitatively similar temporal evolution of the C1–C5 molecules (excluding C2H2) (Fig. S7), C6–C10 (Fig. S8), and > C10 molecules (Fig. S9). The temporal change of the amount of C1–C5, C6–C10, and > C10 species is also shown in terms of molecular percentage by weight (wt %) in Fig. S10.
Figure 4 shows the temporal evolution of the average ratio of (a) C1–C5 molecules including C2H2, (b) C1–C5 molecules excluding C2H2, (c) C6–C10, and (d) > C10 molecules for the acetylene pyrolysis simulations of Fig. 1, at 1350 (circles), 1500 (squares), 1650 (diamonds), and 1800 K (stars). During inception at T = 1350 K (t ≤ 4.5 ns), the average ratio of all C1–C5 molecules is 1 (Fig. 4a) as most (> 95 %) of the reactive species is acetylene (Fig. 3a), while the remaining 5 % exhibits a slightly higher ratio of up to 1.3 (Fig. 4b). After the onset of surface growth (i.e., t > 5.2 ns at 1350 K), the average ratio of C1–C5 molecules slightly decreases, attaining a value of 0.53, including (Fig. 4a) and excluding (Fig. 4b) C2H2, as most acetylene has been depleted (Fig. 3a). As larger molecules start to form (Fig. 3c and d), the ratio of C6–C10 and > C10 species increases, reaching a maximum shortly after surface growth starts (t ≅ 5–6 ns). This attainment of the maximum ratio of intermediate-sized or large molecules coincides with the maximum number concentration of those compounds throughout soot formation. For t ≥ 6.5 ns, where steady > C10 concentration is reached (Fig. 3d), the average ratio of the > C10 molecules is approaching 2 (Fig. 4d). During soot surface growth (t ≥ 5.2 ns), the ratio of C1–C5 molecules drops, reaching a plateau at 0.57 (Fig. 4a) or at 0.41 without accounting for C2H2 (Fig. 4b). The ratio of C6–C10 molecules decreases once the incipient soot is formed (Fig. 4c, t ≥ 6.5 ns) until the attainment of a plateau of about 1. The C6–C10 molecules show a higher ratio than that of the C1–C5 molecules during inception. The peak in the ratio of the C6–C10 molecules coincides with the cluster formation (i.e., when molecular weight of the incipient soot is ∼ 202 g mol−1), indicating that these molecules act as nuclei for the formation of the soot cluster.
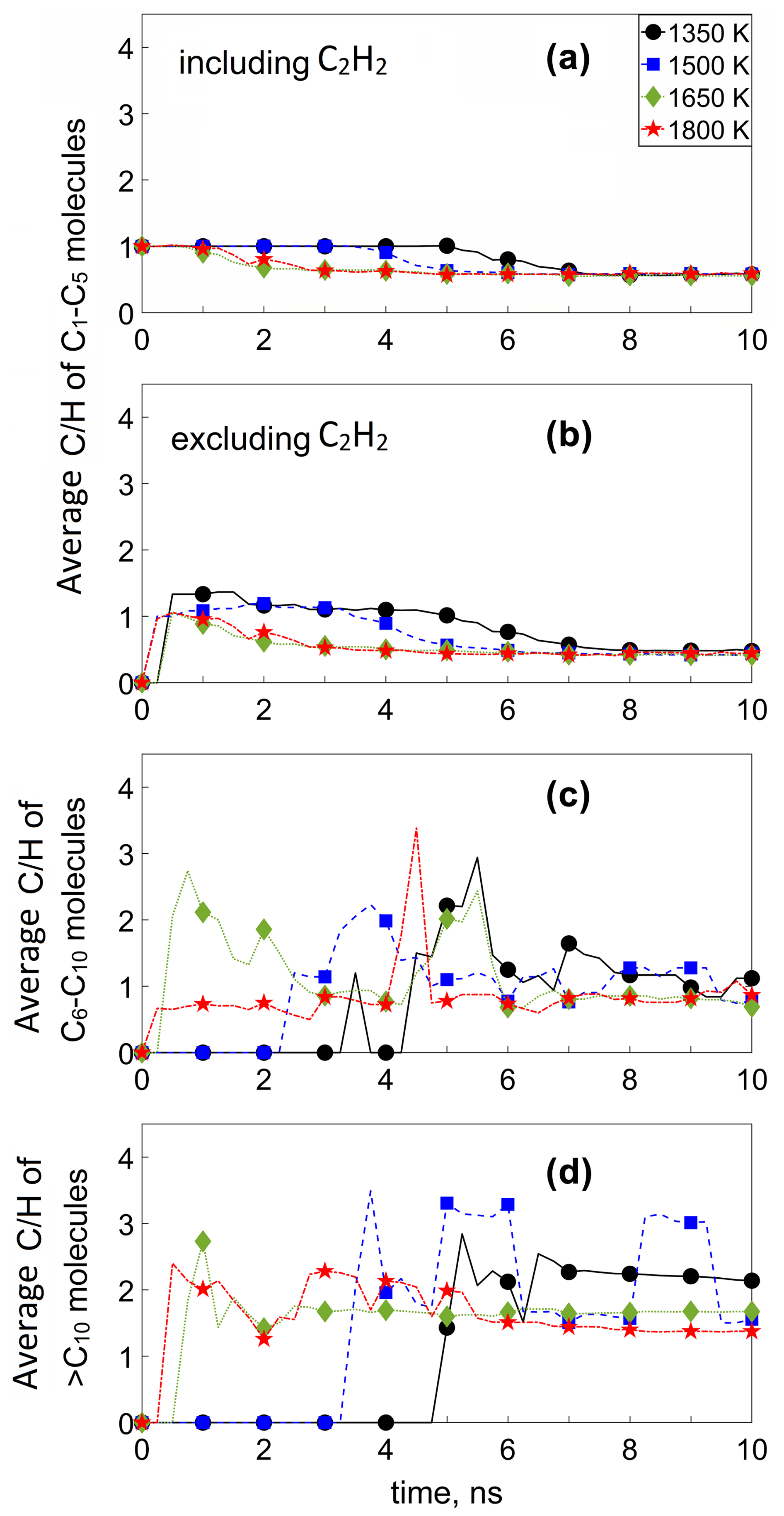
Figure 4Temporal evolution of the average ratio of (a) C1–C5 molecules including C2H2, (b) C1–C5 molecules excluding C2H2, (c) C6–C10, and (d) > C10 molecules for the acetylene pyrolysis simulations of Fig. 1, at 1350 (circles), 1500 (squares), 1650 (diamonds), and 1800 K (stars).
Increasing the process temperature leads to faster formation of C1–C5 molecules, which eventually reach the same ratio of 0.41 during soot surface growth (Fig. 4b). This increase in temperature, however, leads to faster attainment of this asymptotic ratio of small (< C5) molecules. Also, higher temperature (> 1650 K) leads to faster formation of C2H4, CH4, C2H3, and C2H6 (please see also Fig. 5b–e), which reduce the average ratio of C1–C5 molecules until a plateau is reached around 3–4 ns (Fig. 4b). Since no stable > C5 molecules are formed before the incipient soot formation (Figs. 1, 2, and 3c and d), the ratio for C6–C10 molecules is mainly observed post-inception for all temperatures, and only when > C5 molecules are formed does the ratio slightly fluctuate as the cluster grows (Fig. 4c and d). Additionally, the > C10 (Fig. 4d) and C6–C10 (Fig. 4c) molecules are more carbonized than their smaller counterparts (C1–C5 molecules), indicated by their higher ratio. The ratio of the incipient soot during acetylene pyrolysis (Fig. 4d) is lower at higher temperature (≥ 1650 K) due to dehydrogenation, consistent with experiments with other fuel flames (e.g., methane, Alfè et al., 2009, 2010; Russo et al., 2015, 2013; ethylene, Alfè et al., 2009; Russo et al., 2015; Russo et al., 2013). The evolution has been reproduced for up to four MD simulations with various initial configurations (Figs. S11–S14), confirming the trends shown in Fig. 4.
The ratio of the MD-obtained non-cyclic (Fig. S15: diamonds) and cyclic soot precursors (Fig. S15: circles) consisting of 11–70 C atoms formed throughout the nucleation and surface growth stages (t = 0–10 ns) is compared to that of soot precursors obtained during ethylene combustion by high-resolution atomic force microscopy (AFM) measurements (Commodo et al., 2019; Lieske et al., 2023) (Fig. S15: filled symbols) at 1350–1800 K. Most of the soot precursor molecules composed of up to 25 C atoms exhibit a ratio in the range of 1–2, consistent with the ratio of the molecules observed by AFM (Commodo et al., 2019; Lieske et al., 2023), which range between 1 and 2.5. Larger MD-obtained clusters attain a ratio of 1–1.5, contrary to experiments revealing higher ratios of ∼ 2. This can be attributed to the larger fraction of aromatic rings observed in the soot clusters sampled from premixed ethylene (C2H4)/air flames (Commodo et al., 2019; Schulz et al., 2019), along with the fact that side-aliphatic chains had been excluded from the calculation of the ratio of these AFM-obtained soot clusters (Commodo et al., 2019). This difference between AFM measurements (Commodo et al., 2019; Lieske et al., 2023) and MD simulations could also arise from the presence of O2 in experiments that might have led to the formation of different soot precursors than those obtained here by pyrolysis alone. Aliphatic moieties, however, have been observed in AFM mainly in the form of methyl groups or larger alkyl chains (Schulz et al., 2019). Additionally, the characteristics of soot nanoparticles obtained at the end of these simulations have been rigorously validated in Mukut et al. (2024), further confirming the validity of the present simulations.
Figure 5 shows the temporal evolution of the number of (a) C2 and C, (b) C2H4 and C4H2, (c) CH4 and CH3, (d) C2H3 and C2H, and (e) C2H6 and C2H5, corresponding to the 10 most abundant organic species, and (f) H2 and H formed by acetylene pyrolysis during inception and surface growth at 1350 (black lines), 1500 (blue lines), 1650 (green lines), and 1800 K (red lines). The concentration of all molecules increases, while C2H2 is rapidly consumed (Fig. 3a). At the onset of surface growth, C2H3 exhibits the highest concentration (Fig. 5d, solid line), suggesting that the intermediates formed by reactive collisions of acetylene are eventually converted to vinyl radicals, as proposed in the hydrogen abstraction vinyl acetylene addition (HAVA) mechanism (Shukla and Koshi, 2012).
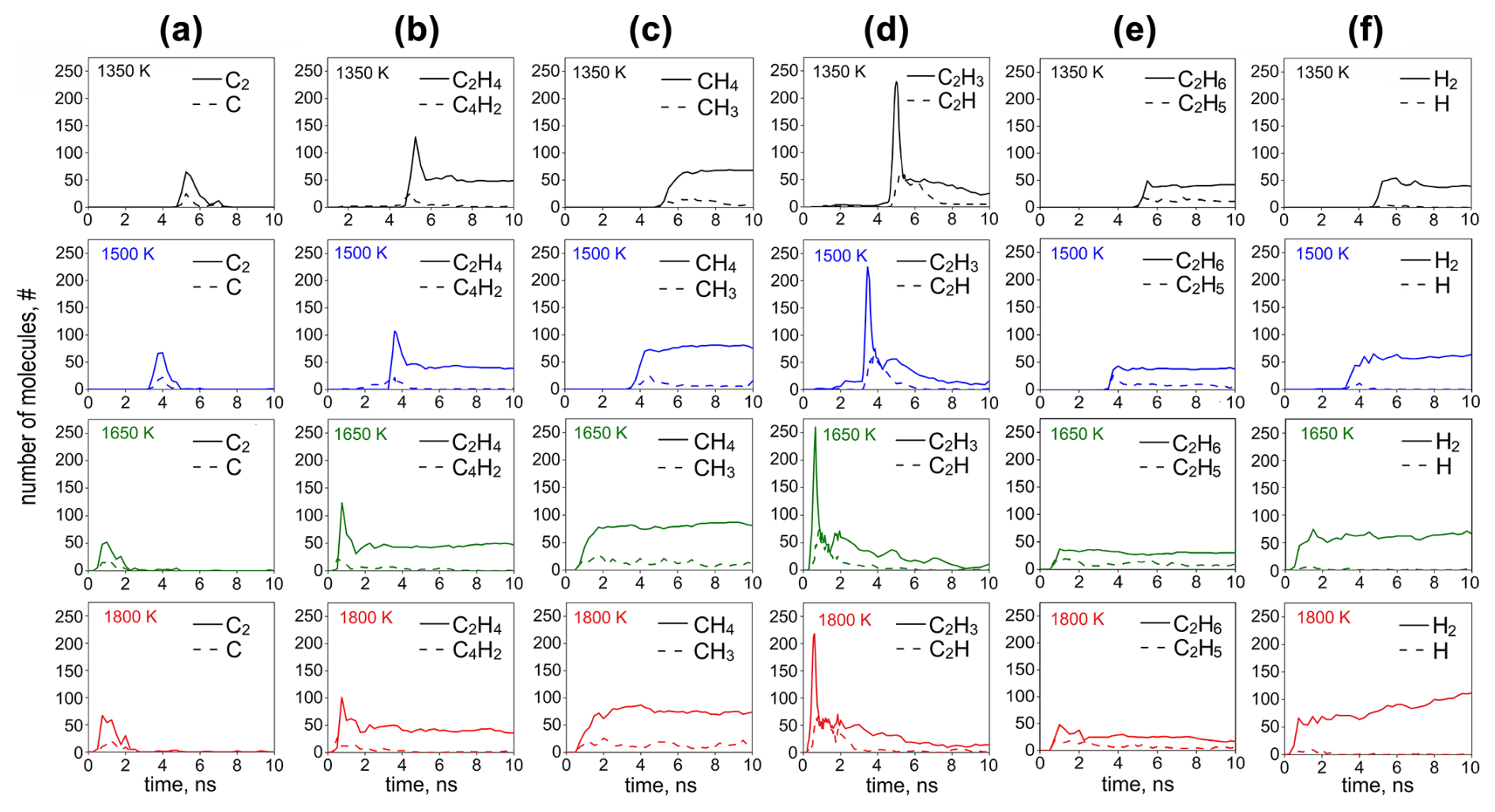
Figure 5Temporal evolution of the number of (a) C2 and C, (b) C2H4 and C4H2, (c) CH4 and CH3, (d) C2H3 and C2H, (e) C2H6 and C2H5, and (f) H2 and H corresponding to the most abundant species formed by acetylene pyrolysis during inception (t = 0–10 ns) at 1350 (black lines), 1500 (blue lines), 1650 (green lines), and 1800 K (red lines).
For example, at T = 1350 K, the C2H3 (Fig. 5d) and C2H4 exhibit a peak (Fig. 5b) at t = 5.25 ns, which corresponds to the time step right after the onset of the surface growth (Fig. 3). There (t = 5.25 ns), only a small amount of C2H (Fig. 5d), C4H2 (Fig. 5b), C2, C (Fig. 5a), and H2 (Fig. 5f) is produced, followed by an increase in CH3 and CH4 (Fig. 5c) at t ≥ 5.5 ns, indicating that the primary products during acetylene pyrolysis (such as C2H3, C2H4) also contribute to the formation of hydrocarbon molecules containing > 5 C. The role of C2H3 has also been highlighted in kinetic mechanisms as a key contributing molecule to the formation of cyclopentadiene through its addition to C4H6, which competes with benzene formation pathways and influences the composition and growth of soot particles (Faravelli et al., 1998). The amount of C2H4 (Fig. 5b: solid lines), CH4 (Fig. 5c: solid lines), C2H6 (Fig. 5e: solid lines), and H2 (Fig. 5f: solid lines) hardly changes at longer times, after C2H2 has practically been depleted, indicating that these species do not contribute to the growth of the soot cluster. The abrupt decrease in C2H4 (Fig. 5b: solid lines), C2H3 (Fig. 5d: solid lines), and C2H concentration (Fig. 5b: broken lines) also hints that these species contribute towards the growth of incipient soot. It should be noted that the contribution of polyynes, such as C4H2 and C2H, has also been recognized in accelerating polymerization reactions that lead to soot nucleation (Indarto, 2008). The process temperature hardly affects the concentration of the organic species, but higher temperatures lead to faster reactions, as discussed in Fig. 3. It is worth noting that at low temperature (T = 1350–1650 K), dehydrogenation only takes place right after the onset of surface growth, and the H2 concentration remains constant at longer times. At T = 1800 K, however, even though dehydrogenation starts at the onset of surface growth, coinciding with the formation of C6–C10 molecules (Fig. 3c), it continues throughout surface growth of the incipient soot as H2 increases for t ≥ 4 ns (Fig. 5f: T = 1800 K, solid line). The large H2 concentrations generated by pyrolysis at high T are consistent with the H2 synthesis in plasma reactors for carbon black production (Fulcheri and Schwob, 1995).
Figure 6 shows the temporal evolution of the number of (a) three-, (b) five-, and (c) six-member C rings across all molecules during acetylene pyrolysis at 1350 (circles), 1500 (squares), 1650 (diamonds), and 1800 K (stars), with four- and seven-member rings shown in Fig. S16. The appearance of the three-member rings occurs at t = 3.75 ns at 1350 K after C2H2 has practically been depleted (Fig. 3a) and coincides with the formation of C6–C10 molecules (Fig. 3c), indicating the presence of three-member rings in molecules consisting of 6–10 C atoms. The total number of three-member rings decreases when small molecules are consumed and the incipient soot grows, revealing that three-member rings are thermodynamically less stable (Kim and Ihee, 2012; Fantuzzi et al., 2013) compared to the acyclic small molecules (C1–C5) and dissociate when surface growth prevails. The five- and six-member rings are formed simultaneously during pyrolysis. Early on, before the onset of surface growth, no five-, six-, and seven-member rings are observed (Figs. 6 and S16), indicating that these rings mainly exist in the incipient soot. For example, at T = 1350 K (Fig. 6c), six-member rings start to form at t = 6 ns after more than 60 % of the C6–C10 molecules have been depleted (Fig. 3c: t = 6 ns). At longer times, where incipient soot cluster growth steadies, the concentration of five- and six-member rings reaches a plateau, corroborating the fact that these rings originate from the formation of the incipient soot.
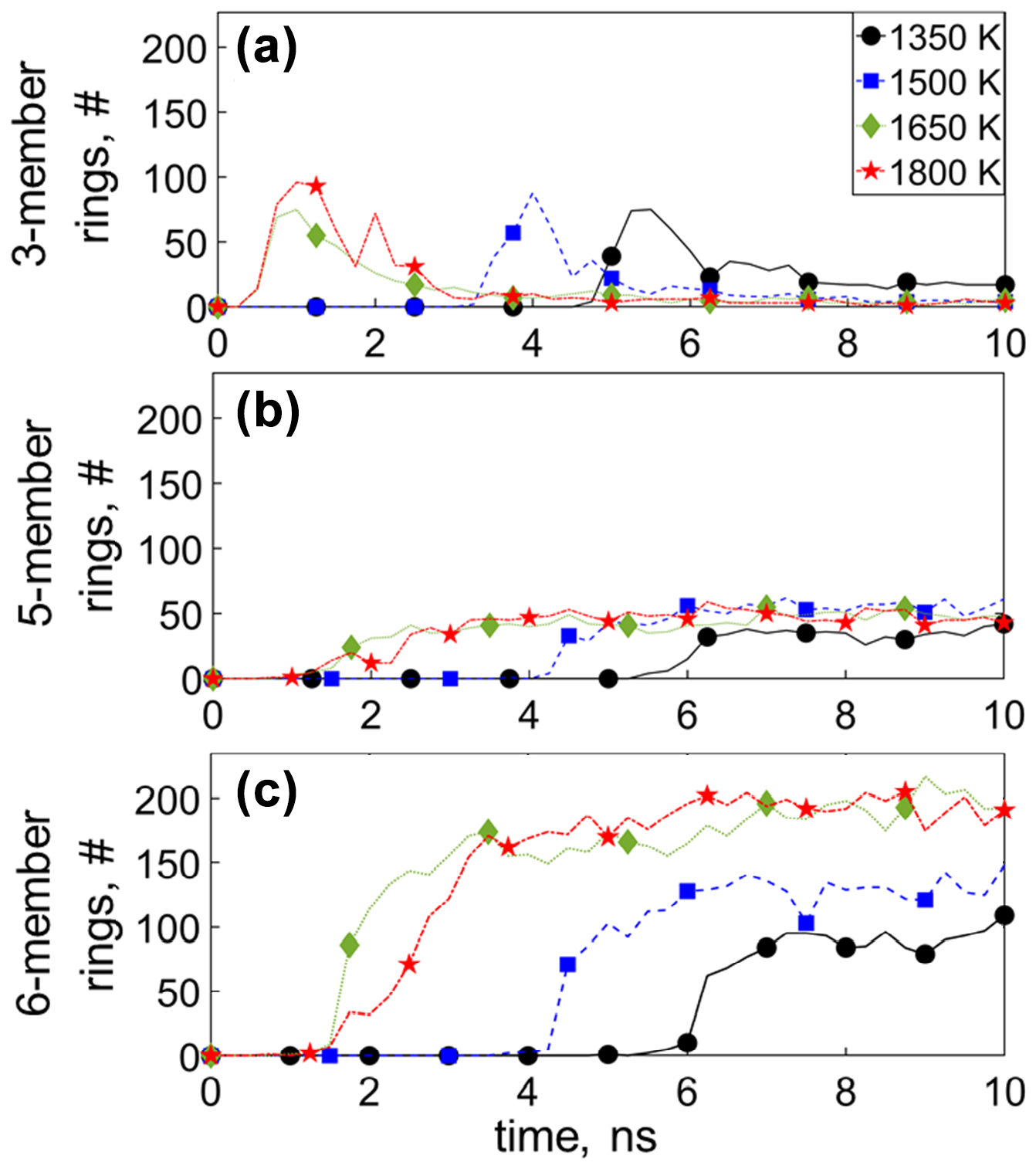
Figure 6Temporal evolution of the number of (a) three-, (b) five-, and (c) six-member C rings during acetylene pyrolysis at 1350 (circles), 1500 (squares), 1650 (diamonds), and 1800 K (stars). The appearance of the three-member rings occurs after C2H2 has been depleted. The total number of three-member rings decreases when small molecules are consumed, and the incipient soot grows while the number of five- and six-member rings increases.
The formation of five- and six-member rings is tracked exemplarily in Fig. S17 for different molecules at T = 1500 K. The initial small hydrocarbons polymerize by reactive collisions, forming extended aliphatic chains consisting of more than 6 C atoms. These large aliphatic chains undergo molecular rearrangement and cyclization, resulting in the formation of five-member and six-member aromatic rings (e.g., Fig. S17a: t = 7.55 and 7.75 ns). These rings dynamically form and may dissociate; for instance, the rings highlighted in Fig. S17a persist for approximately 0.25 ns before dissociating due to collisions with other molecules. Concurrently, other large aliphatic molecules coalesce with existing rings, stabilizing the incipient soot nanoparticle (Fig. S17b: t = 9.2). This leads to further ring formation through molecular rearrangement, rather than through PAH dimerization, thereby underscoring the complex and transient nature of soot precursor dynamics at high temperature. The present ReaxFF simulations can capture the formation of benzene, a major intermediate of soot formation (Fig. S17b, bottom), consistent with reaction mechanisms of acetylene pyrolysis (Saggese et al., 2014).
At the onset of surface growth, the five-member aromatic rings are approximately 13 % for 1350 K and 15 % for 1500, 1650 and 1800 K of the total number (aromatic and aliphatic) of the five-member rings formed (Fig. S18a). At longer times, the fraction of five-member aromatic rings drops down to 3 % for 1350 K and 2 % for 1500, 1650 and 1800 K (Fig. S18a), while the fraction of six-member aromatic rings is 6 % for 1350 and 1650 K and 5 % for 1500 and 1800 K (Fig. S18b). As time proceeds, the fraction of six-member aromatic rings rises to 12 % and 10 % for 1350 and 1500 K and 11 % for both 1650 and 1800 K, consistent with recent MD results (Han et al., 2017). In addition, the total number of six-member rings increases with increasing temperature (Fig. 6c), and cyclization within the incipient soot takes place.
This progression of ring formation is reflected in the size (Fig. S19) and molecular weight distributions (Fig. S20) of the population of all species during soot inception. At 1350 K for t ≤ 3.75 ns and at 1500 K for t ≤ 2 ns, more than 95 % of molecules are acetylene, so there is hardly any change in the molecular weight distribution, and only a few of them have molecular weights up to 200 g mol−1, in line with Figs. 1 and 3a. At longer times, i.e., at 1350 K and t ≥ 5 ns (Figs. S19a and S20a) and at 1500 K and t ≥ 3.75 ns (Figs. S19b and S20b), the distributions shift to larger cluster sizes and molecular weights, with the formation of molecules, radicals, and even a nascent soot cluster having molecular weight between 200 and 1000 g mol−1. The peak corresponding to the largest cluster shifts towards 10 000 g mol−1 for t ≥ 6 ns at 1350 K (Fig. S20a) and for t ≥ 5 ns at 1500 K (Fig. S20b). Likewise, at 0.75 ns for 1650 (Fig. S20c) and 1800 K (Fig. S20d), the molecular weight distribution of reactive components spans 100 to 1000 g mol−1, indicating that nucleation has practically stopped, and nascent soot clusters have been formed. At 6 ns and beyond, the growth of the soot cluster stops (with molecular weight peaks being observed around 10 000 g mol−1) due to depletion of the reactive species.
3.2 Chemical structure of soot precursors
Tables 2–5 show a breakdown of the detailed chemical structure of the molecules generated during acetylene pyrolysis at 1500 K for 0.75 and 2 (Table 2), 3.75 (Table 3), 5 and 6 (Table 4), and 8 ns (Table 5) for the simulation shown in Fig. 1b. For clarity, the incipient soot is omitted at 5, 6, and 8 ns.
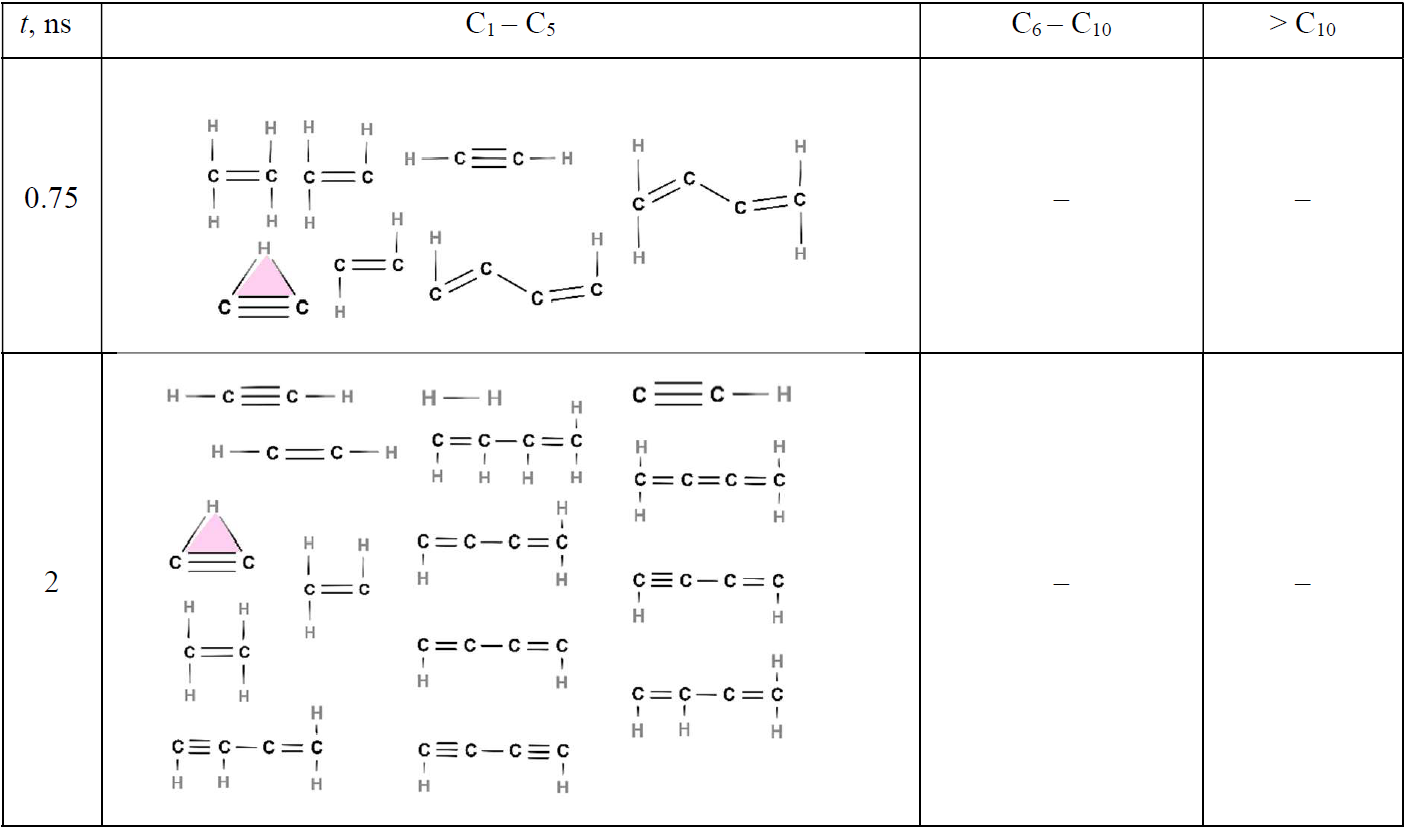
Early on (t ≤ 2 ns; Table 2), only small molecules consisting of up to 4 C atoms are observed. Among those, acetylene is the most abundant (Fig. 3a), while H2, C2H (cyclic and linear), C2H3, C2H4, C4H2 (as shown in Fig. 5), C4H3, and C4H4 are also present at low concentrations. Shortly after the onset of surface growth, at t = 3.75 ns (Tables 3 and S2), two (C84H39 and C87H40) soot nuclei appear (Table S2). Parallel to this, both cyclic and linear molecules with more than 6 C atoms emerge with most of the cyclic structures consisting of three-member rings (highlighted in yellow). Some of these three-member rings reported in Table 3 correspond to known monocyclic molecules, including cyclopropyne (Saxe and Schaefer, 1980) (molecule A); bicyclic molecules, including bicyclo(1.1.0)butane (Fawcett, 2020) (molecule B); cyclopropane (Li et al., 2020) (molecule C); spiropentadiene (Billups and Haley, 1991) (molecule D); and cyclopropylidene cyclopropane (Güney et al., 2013) (molecule E). It should be noted that some of the three-member ring structures predicted by ReaxFF, such as cyclopropyne, are not inherently stable and may exist only transiently in high-energy states. C2H cyclic molecules (highlighted in pink) having three-center two-electron configuration (Lammertsma and Ohwada, 1996) are observed at all six time steps. Cyclobutane (highlighted in green) is only detected at 3.75 ns (Table 3), indicating its thermodynamic instability, consistent with Zádor et al. (2017). Aliphatic compounds such as propane (Iijima, 1972); propene (Lide and Christensen, 1961); butane (Bradford et al., 1977); butene (Lu et al., 2017); and other alkanes, alkenes, and alkynes are also observed at 3.75 ns or later (Tables 3–5). The five- (blue-shaded rings) and six-member (red-shaded rings) rings typically manifest in > C10 molecules (Tables 3 and S2). The small molecules (H2, C2H, C2H2, C2H3, C2H4, C4H2, C4H3, and C4H4) that have formed at earlier stages, persist at 3.75, 5, and 6 ns (Tables 3 and 4), albeit with a notable reduction in their number concentration.
Table 2Chemical structures of all molecules formed during acetylene pyrolysis at t = 0.75 and 2 ns at 1500 K. Molecules larger than C4 are not formed within the first 2 ns of acetylene pyrolysis at this temperature.
Due to their thermodynamic instability, the three-member rings formed early on (e.g., Table 3: t = 3.75 ns) dissociate almost immediately, as indicated by the drop in their number at 5, 6, and 8 ns, while molecules other than the incipient soot possess only a few five- or six-member rings during these time steps (Tables 4 and 5). However, the total number of five- or six-member rings in the entire population of soot precursor molecules is much higher (approximately 50 five-member rings, Fig. 6b, and 100–150 six-member rings, Fig. 6c), suggesting that almost all of them belong to the incipient soot.
Table 3Chemical structures of all molecules, composed of up to 25 carbon atoms, formed during acetylene pyrolysis at 3.75 ns at 1500 K. Two larger nuclei (C84H39 and C87H40) are also present (shown in Table S2).
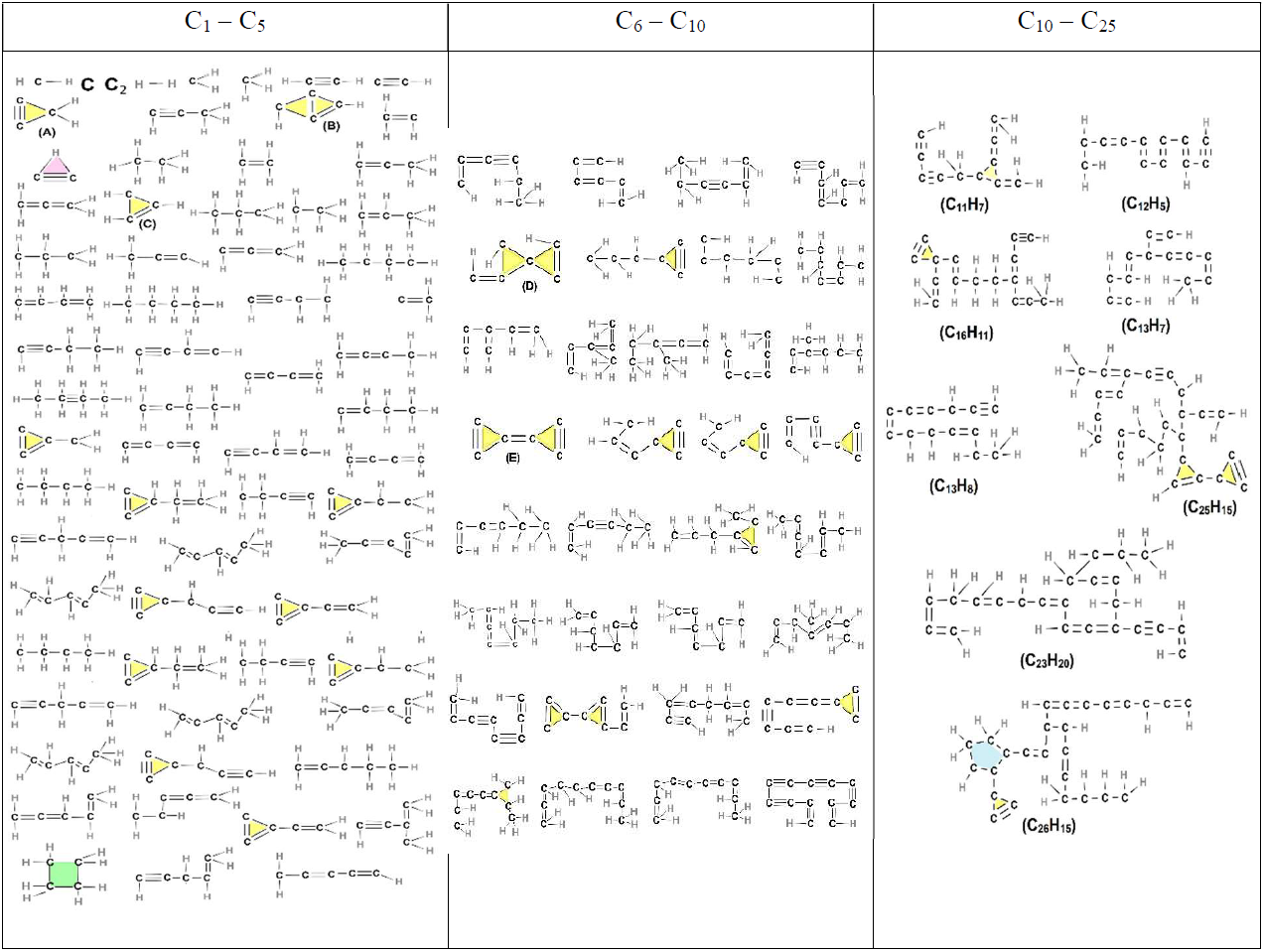
Table 4List of all molecules, aliphatic, cyclic, or aromatic, except the incipient soot, formed during acetylene pyrolysis at 5 (top row) and 6 ns (bottom row) at 1500 K, including their chemical structures. The excluded incipient soot consists of 1266 and 1307 C atoms at 5 and 6 ns, respectively.
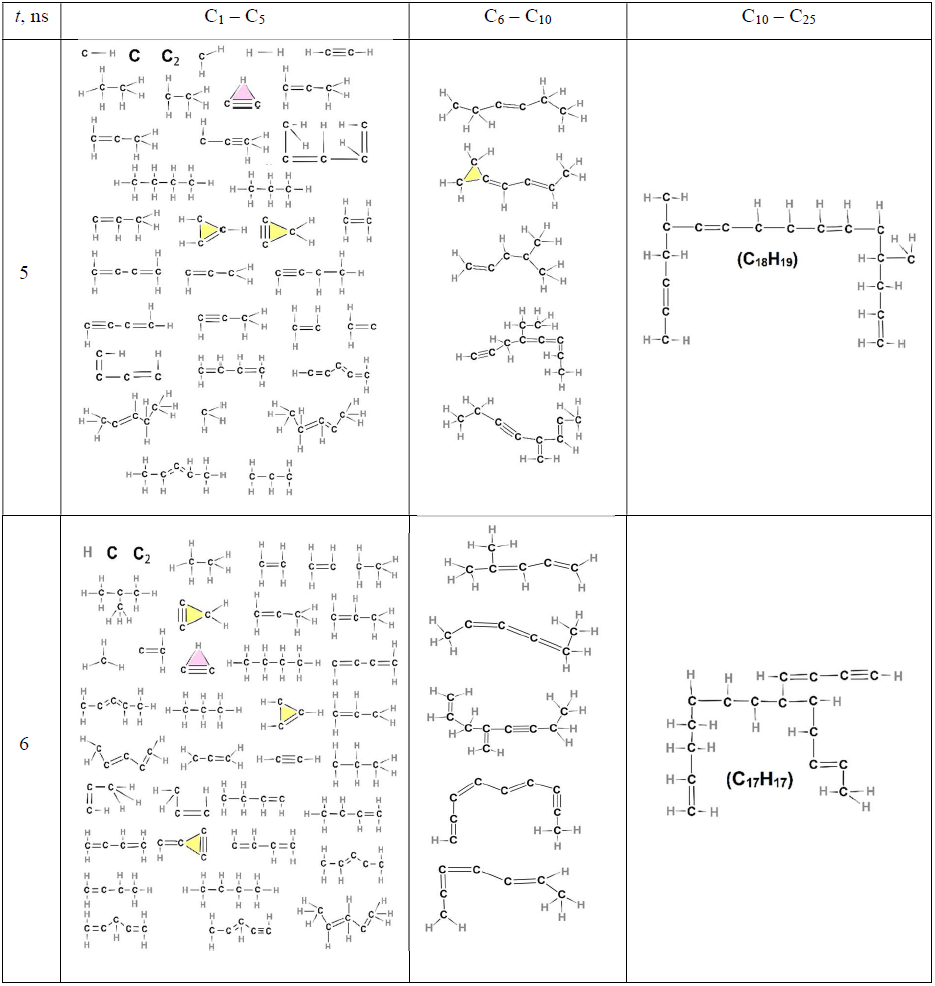
Table 5List of all molecules, aliphatic, cyclic, or aromatic molecules, except the incipient soot (consisting of 1335 C atoms) formed during acetylene pyrolysis at 8 ns at 1500 K, including their chemical structures.
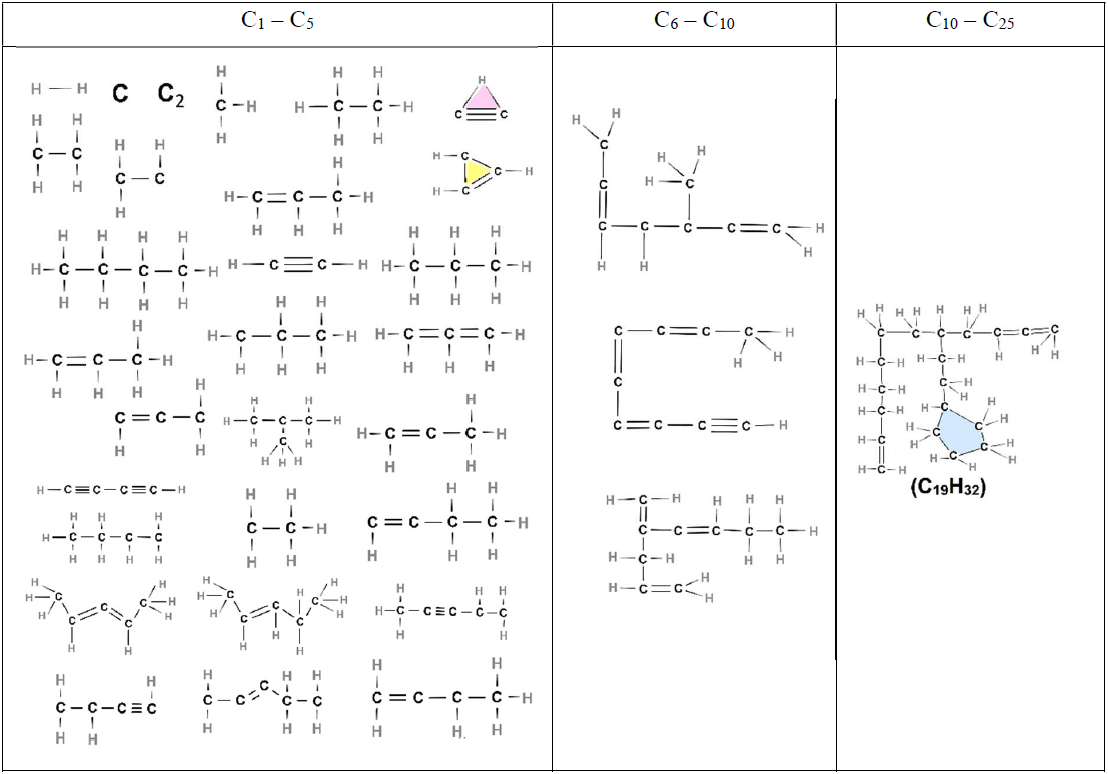
At the initial stages of soot inception (t = 0.75 and 2 ns), linear and branched aliphatic hydrocarbons are formed along with unsaturated carbon chains and radical sites. One of the most abundant radicals formed is ethenyl (C2H3, Fig. 5d). Due to its rapid formation and almost immediate consumption, ethenyl is crucial for further growth of hydrocarbon structures and soot inception, consistent with Wang et al. (2021). By adding to unsaturated carbon bonds, ethenyl can facilitate chain propagation and cross-linking for cyclization, leading to the formation of new rings, as evidenced by C26H15 molecules at 3.25 ns (Table 3) and C19H32 molecules at 8 ns (Table 5).
The present results highlight the relevance of chemical nucleation in soot formation at intermediate temperatures (1350–1800 K), emphasizing a substantial role for chemical pathways even in the absence of observable physical PAH dimerization. This is in stark contrast to ReaxFF simulations that start with PAHs as monomers (Mao et al., 2017), indicating lack of nucleation at temperatures ranging from 1200 to 2400 K, despite soot yield peaks within this temperature range (Frenklach et al., 1983).
The detailed chemical structure of molecules formed during acetylene pyrolysis is also shown for 1350, 1650 and 1800 K at 0.75, 2, 3.75, 5, 6, and 8 ns (Tables S3–S15 in the Supplement). For t ≤ 3.75 ns, formation of vinyl molecules (C2H3) and cyclic C2H molecules is observed along with small aliphatic molecules (< 5 C atoms) at 1350 K (Table S3). For 1350 K at t = 5 ns (Table S4), three-member rings and molecules containing > 5 C atoms are formed, coinciding with C2H2 consumption (Fig. 3a). At this temperature, hardly any five- and six-member rings are found in the reported chemical structures (Tables S3–S6), which exclude the incipient soot, indicating that most of these rings are formed within the incipient soot nanoparticle. Nevertheless, at t = 5 ns (i.e., 0.2 ns prior to the onset of surface growth), a molecule with a six-member ring appears in the C6–C10 range (Table S4), but most of the larger molecules with > C10 are primarily long aliphatic chains.
Smaller benzene derivatives are observed in the reaction pathway at 1650 (Tables S10–S11) and 1800 K (Tables S14–S15). At both 1650 (Tables S7–S11) and 1800 K (Tables S12–S15), mostly three-member rings are formed up to 6 ns, along with the incipient soot (excluded in the tables). Approximately 0.05 ns prior to the onset of surface growth (Table S12: t = 0.75 ns), a five-member ring spontaneously forms. At T = 1650 K, a benzene derivative forms at 5 ns, while naphthalene derivatives form at 6 and 8 ns (Table S11). However, only a few small and intermediate molecules with five- and six-member rings exist at t = 6 and 8 ns at 1800 K (Table S15), further indicating that most of these rings belong to the large incipient soot nanoparticle. At all temperatures employed here (1350–1800 K), large molecules with more than 10 C atoms are composed of a few (five- and six-member rings) decorated with long branches (Tables 3–5, S2, S3–S6, S7–S11, and S12–S15). Even though these structures are different from the majority of those observed experimentally (Lieske et al., 2023; Commodo et al., 2019; Jacobson et al., 2020; Martin et al., 2021), which are composed of aromatic islands with peripheral methyl groups, long alkyl chains have also been observed (Schulz et al., 2019) on par with the present simulations. It should also be noted that at temperatures below 1800 K, which are relevant to soot formation in flames, the ReaxFF-predicted cluster structures are significantly different from those formed by acetylene combustion at 2700 K (Wang et al., 2022a), by methane and ethylene combustion at 3000 K (Wang et al., 2022b), by dimerization of PAHs (Zhao et al., 2020), and by pyrolysis of n-decane at 3000 K (Liu et al., 2020) or n-heptane at 2200–2600 K (Fakharnezhad et al., 2025), where PAH-like soot precursors are formed, leading to large carbonaceous clusters mainly composed of rings and containing only a small fraction of branches. This indicates that the fuel type along with the nucleation conditions can significantly affect the formation mechanism and structure of soot.
The inception and early stages of soot surface growth by acetylene pyrolysis are investigated using reactive molecular dynamics at 1350–1800 K. These simulations do not assume any specific soot precursors, allowing for a broader exploration of the pathways involved in soot nucleation. Increasing the temperature leads to faster formation of the incipient soot taking place through linearization, cyclization, and subsequent surface condensation of radicals on the incipient soot. During linearization and cyclization, small molecules consisting of fewer than 6 C atoms, such as C2H3, C2H4, C2H6, CH4, and C2, are formed at all temperatures. These small C3–C6 molecules are mainly aliphatic chains or three-member rings. Increasing the process temperature leads to faster depletion of C2H2 molecules and faster formation of these compounds. The growth of small C1–C5 molecules can be attributed to reactive collisions, which eventually lead to the formation of larger aliphatic compounds consisting of 6–10 C atoms. At the initial stages of inception and prior to the formation of the incipient soot, three-member rings are formed, associated with the formation of compounds with fewer than 10 C atoms. Once the incipient soot is formed, the number of C1–C10 compounds and three-member rings drops, while the number of five- and six-member rings increases, indicating that the formation of larger rings is associated with the formation and growth of the incipient soot. The cyclic structures are mainly observed within the incipient soot, which is supported by the information of the detailed chemical structures of the molecules observed at different time steps. Most of these cyclic structures consist of a few rings interconnected with aliphatic side chains. By providing a comprehensive list of all molecular precursors – rather than focusing solely on the most abundant ones – this study offers a more complete view of the chemical complexity involved in soot nucleation. Tracking the pathway of formation of these species could reveal new detailed chemical routes that occur during soot nucleation, which might have not been extensively considered in existing kinetic models, thereby expanding the current understanding of soot formation pathways. However, verification of ReaxFF-derived reactions with ab initio quantum mechanics calculations, such as density functional theory, is essential to ensure the importance of these reactions in soot formation kinetics.
The open-source software LAMMPS (https://www.lammps.org/download.html; Plimpton, 1995) was used for running the reactive molecular dynamics simulations. Post-processing of the chemical structures and ring analysis were carried out as documented in Sect. 2, using the Chemical Trajectory Analyzer (ChemTrayZer) package (https://github.com/yiming-xu/LAMMPS_Simulation/tree/master/ChemTraYzer_2.0/py3; Döntgen et al., 2018) and the MAFIA-MD software (https://github.com/comp-comb/MAFIA-MD; Mukut et al., 2022), respectively.
The trajectories and bond order information files generated by the molecular dynamics simulations can be found in the Zenodo repository: https://doi.org/10.5281/zenodo.15162017 (Ganguly, 2025).
Figures showing the onset of incipient soot, the temporal evolution of acetylene and other hydrocarbons, the composition ( ratio), the five- and six-member aromatic rings, the size distribution of molecules as a function of volume-equivalent diameter, and the molecular weight distribution of molecules at 1350–1800 K, as well as tables showing the incipient soot for all simulations and the chemical structures of the hydrocarbons at various time steps at 1350, 1650, and 1800 K during acetylene pyrolysis, are provided. The supplement related to this article is available online at https://doi.org/10.5194/ar-3-185-2025-supplement.
AG: writing (original draft), visualization, validation, software, methodology, formal analysis, data curation, conceptualization. KMM: writing (review and editing), methodology. SR: writing (review and editing), methodology, supervision, resources. GAK: writing (review and editing), methodology. EG: writing (review and editing), supervision, software, resources, methodology, conceptualization.
At least one of the (co-)authors is a member of the editorial board of Aerosol Research. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The research benefited from computational resources provided through the National Computational Merit Allocation Scheme, supported by the Australian Government, and the University of Melbourne's Research Computing Services and the Petascale Campus Initiative. Anindya Ganguly acknowledges Felix Schmalz for his assistance with ChemTraYzer software. Khaled Mosharraf Mukut and Somesh Roy acknowledge funding support from the National Science Foundation as some of this material is based upon work supported by the National Science Foundation under grant no. 2144290.
This research has been supported by the National Science Foundation (grant no. 2144290).
This paper was edited by Jose Castillo and reviewed by two anonymous referees.
Agafonov, G. L., Bilera, I. V., Vlasov, P. A., Kolbanovskii, Y. A., Smirnov, V. N., and Tereza, A. M.: Soot formation during the pyrolysis and oxidation of acetylene and ethylene in shock waves, Kinet. Catal., 56, 12–30, https://doi.org/10.1134/S0023158415010012, 2015.
Aghsaee, M., Dürrstein, S. H., Herzler, J., Böhm, H., Fikri, M., and Schulz, C.: Influence of molecular hydrogen on acetylene pyrolysis: Experiment and modeling, Combust. Flame, 161, 2263–2269, https://doi.org/10.1016/j.combustflame.2014.03.012, 2014.
Alfè, M., Apicella, B., Barbella, R., Rouzaud, J. N., Tregrossi, A., and Ciajolo, A.: Structure–property relationship in nanostructures of young and mature soot in premixed flames, P. Combust. Inst., 32, 697–704, https://doi.org/10.1016/j.proci.2008.06.193, 2009.
Alfè, M., Apicella, B., Rouzaud, J. N., Tregrossi, A., and Ciajolo, A.: The effect of temperature on soot properties in premixed methane flames, Combust. Flame, 157, 1959–1965, https://doi.org/10.1016/j.combustflame.2010.02.007, 2010.
Anenberg, S. C., Schwartz, J., Shindell, D., Amann, M., Faluvegi, G., Klimont, Z., Janssens-Maenhout, G., Pozzoli, L., Van Dingenen, R., Vignati, E., Emberson, L., Z, M. N., Jason, W. J., Williams, M., Demkine, V., Kevin, H. W., Kuylenstierna, J., Raes, F., and Ramanathan, V.: Global Air Quality and Health Co-benefits of Mitigating Near-Term Climate Change through Methane and Black Carbon Emission Controls, Environ. Health Persp., 120, 831–839, https://doi.org/10.1289/ehp.1104301, 2012.
Appel, J., Bockhorn, H., and Frenklach, M.: Kinetic modeling of soot formation with detailed chemistry and physics: laminar premixed flames of C2 hydrocarbons, Combust. Flame, 121, 122–136, https://doi.org/10.1016/S0010-2180(99)00135-2, 2000.
Arvelos, S., Abrahão, O., and Eponina Hori, C.: ReaxFF molecular dynamics study on the pyrolysis process of cyclohexanone, J. Anal. Appl. Pyrol., 141, 104620, https://doi.org/10.1016/j.jaap.2019.05.009, 2019.
Bergwerf, H.: MolView: an attempt to get the cloud into chemistry classrooms, DivCHED CCCE: Committee on Computers in Chemical Education, 9, 1–9, 2015.
Billups, W. E. and Haley, M. M.: Spiropentadiene, J. Am. Chem. Soc., 113, 5084–5085, https://doi.org/10.1021/ja00013a067, 1991.
Bond, T. C., Doherty, S. J., Fahey, D. W., Forster, P. M., Berntsen, T., DeAngelo, B. J., Flanner, M. G., Ghan, S., Kärcher, B., Koch, D., Kinne, S., Kondo, Y., Quinn, P. K., Sarofim, M. C., Schultz, M. G., Schulz, M., Venkataraman, C., Zhang, H., Zhang, S., Bellouin, N., Guttikunda, S. K., Hopke, P. K., Jacobson, M. Z., Kaiser, J. W., Klimont, Z., Lohmann, U., Schwarz, J. P., Shindell, D., Storelvmo, T., Warren, S. G., and Zender, C. S.: Bounding the role of black carbon in the climate system: A scientific assessment, J. Geophys. Res.-Atmos., 118, 5380–5552, https://doi.org/10.1002/jgrd.50171, 2013.
Bradford, W. F., Fitzwater, S., and Bartell, L. S.: Molecular structure of n-butane: calculation of vibrational shrinkages and an electron diffraction re-investigation, J. Mol. Struct., 38, 185–194, https://doi.org/10.1016/0022-2860(77)87091-9, 1977.
Castro-Marcano, F., Kamat, A. M., Russo, M. F., van Duin, A. C. T., and Mathews, J. P.: Combustion of an Illinois No. 6 coal char simulated using an atomistic char representation and the ReaxFF reactive force field, Combust. Flame, 159, 1272–1285, https://doi.org/10.1016/j.combustflame.2011.10.022, 2012.
Chenoweth, K., van Duin, A. C. T., and Goddard, W. A.: ReaxFF Reactive Force Field for Molecular Dynamics Simulations of Hydrocarbon Oxidation, The J. Phys. Chem. A, 112, 1040–1053, https://doi.org/10.1021/jp709896w, 2008.
Chenoweth, K., van Duin, A. C. T., Dasgupta, S., and Goddard Iii, W. A.: Initiation Mechanisms and Kinetics of Pyrolysis and Combustion of JP-10 Hydrocarbon Jet Fuel, J. Phys. Chem. A, 113, 1740–1746, https://doi.org/10.1021/jp8081479, 2009.
Commodo, M., Kaiser, K., De Falco, G., Minutolo, P., Schulz, F., D'Anna, A., and Gross, L.: On the early stages of soot formation: Molecular structure elucidation by high-resolution atomic force microscopy, Combust. Flame, 205, 154–164, https://doi.org/10.1016/j.combustflame.2019.03.042, 2019.
Dewa, K., Ono, K., Watanabe, A., Takahashi, K., Matsukawa, Y., Saito, Y., Matsushita, Y., Aoki, H., Era, K., Aoki, T., and Yamaguchi, T.: Evolution of size distribution and morphology of carbon nanoparticles during ethylene pyrolysis, Combust. Flame, 163, 115–121, https://doi.org/10.1016/j.combustflame.2015.09.007, 2016.
Dillstrom, T. and Violi, A.: The effect of reaction mechanisms on the formation of soot precursors in flames, Combust. Theor. Model., 21, 23–34, https://doi.org/10.1080/13647830.2016.1211741, 2017.
Döntgen, M., Schmalz, F., Kopp, W. A., Kröger, L. C., and Leonhard, K.: Automated Chemical Kinetic Modeling via Hybrid Reactive Molecular Dynamics and Quantum Chemistry Simulations, J. Chem. Inf. Model., 58, 1343–1355, https://doi.org/10.1021/acs.jcim.8b00078, 2018.
Döntgen, M., Przybylski-Freund, M.-D., Kröger, L. C., Kopp, W. A., Ismail, A. E., and Leonhard, K.: Automated Discovery of Reaction Pathways, Rate Constants, and Transition States Using Reactive Molecular Dynamics Simulations, J. Chem. Theory Comput., 11, 2517–2524, https://doi.org/10.1021/acs.jctc.5b00201, 2015 (code available at: https://github.com/yiming-xu/LAMMPS_Simulation/tree/master/ChemTraYzer_2.0/py3, last access: February 2025).
Drakon, A. V., Eremin, A. V., Gurentsov, E. V., Mikheyeva, E. Y., and Kolotushkin, R. N.: Optical properties and structure of acetylene flame soot, Appl. Phys. B, 127, 81, https://doi.org/10.1007/s00340-021-07623-8, 2021.
Emri, J. and Lente, G.: Use of an electron equivalent relationship between bond length and bond order to study chemical bonding. Part II. A study of bond orders, bond lengths and aromaticity in polycyclic aromatic hydrocarbons, J. Mol. Struc.-THEOCHEM, 671, 211–219, https://doi.org/10.1016/j.theochem.2003.11.002, 2004.
Evans, D. J. and Holian, B. L.: The Nose–Hoover thermostat, J. Chem. Phys., 83, 4069–4074, https://doi.org/10.1063/1.449071, 1985.
Fakharnezhad, A., Saad, D. M., Kelesidis, G. A., and Goudeli, E.: Nucleation Rate of Soot by n-Heptane Pyrolysis, Aerosol Sci. Tech., in press, https://doi.org/10.1080/02786826.2025.2480625, 2025.
Fantuzzi, F., Baptista, L., Rocha, A. B., and da Silveira, E. F.: Theoretical and experimental investigation on the stability of CH− and CH clusters, Chem. Phys., 410, 109–117, https://doi.org/10.1016/j.chemphys.2012.11.010, 2013.
Faravelli, T., Goldaniga, A., and Ranzi, E.: The kinetic modeling of soot precursors in ethylene flames, Symposium (International) on Combustion, 27, 1489–1495, https://doi.org/10.1016/S0082-0784(98)80556-0, 1998.
Fawcett, A.: Recent advances in the chemistry of bicyclo- and 1-azabicyclo[1.1.0]butanes, Pure Appl. Chem., 92, 751–765, https://doi.org/10.1515/pac-2019-1007, 2020.
Frenklach, M.: Reaction mechanism of soot formation in flames, Phys. Chem. Chem. Phys., 4, 2028–2037, https://doi.org/10.1039/B110045A, 2002.
Frenklach, M. and Wang, H.: Detailed modeling of soot particle nucleation and growth, Symposium (International) on Combustion, 23, 1559–1566, https://doi.org/10.1016/S0082-0784(06)80426-1, 1991.
Frenklach, M. and Wang, H.: Detailed Mechanism and Modeling of Soot Particle Formation, in: Soot Formation in Combustion: Mechanisms and Models, edited by: Bockhorn, H., Springer Berlin Heidelberg, Berlin, Heidelberg, 59, 165–192, https://doi.org/10.1007/978-3-642-85167-4_10, 1994.
Frenklach, M., Taki, S., Durgaprasad, M. B., and Matula, R. A.: Soot formation in shock-tube pyrolysis of acetylene, allene, and 1,3-butadiene, Combust. Flame, 54, 81–101, https://doi.org/10.1016/0010-2180(83)90024-X, 1983.
Fulcheri, L. and Schwob, Y.: From methane to hydrogen, carbon black and water, Int. J. Hydrogen Energ., 20, 197–202, https://doi.org/10.1016/0360-3199(94)E0022-Q, 1995.
Ganguly, A.: Investigation of soot precursor molecules during inception by acetylene pyrolysis using reactive molecular dynamics, Version v1, Zenodo [data set], https://doi.org/10.5281/zenodo.15162017, 2025.
Gao, H. and Tang, H.: Temperature Effect on Formation of Polycyclic Aromatic Hydrocarbons in Acetylene Pyrolysis, ChemistrySelect, 7, e202201893, https://doi.org/10.1002/slct.202201893, 2022.
Giannadaki, D., Lelieveld, J., and Pozzer, A.: Implementing the US air quality standard for PM2.5 worldwide can prevent millions of premature deaths per year, Environmental Health, 15, 88, https://doi.org/10.1186/s12940-016-0170-8, 2016.
Goudeli, E.: Nanoparticle growth, coalescence, and phase change in the gas-phase by molecular dynamics, Curr. Opin. Chem. Eng., 23, 155–163, https://doi.org/10.1016/j.coche.2019.04.001, 2019.
Güney, M., Eşsiz, S., Daştan, A., Balci, M., De Lucchi, O., Şahin, E., and Fabris, F.: Stereoconvergent Generation of a Contrasteric syn-Bicyclopropylidene (=syn-Cyclopropylidenecyclopropane) by Stille-Like Coupling, Helv. Chim. Acta, 96, 941–950, https://doi.org/10.1002/hlca.201200291, 2013.
Han, S., Li, X., Nie, F., Zheng, M., Liu, X., and Guo, L.: Revealing the Initial Chemistry of Soot Nanoparticle Formation by ReaxFF Molecular Dynamics Simulations, Energ. Fuels, 31, 8434–8444, https://doi.org/10.1021/acs.energyfuels.7b01194, 2017.
Hermann, M. and Frenking, G.: The Chemical Bond in C2, Chem.-Eur. J., 22, 4100–4108, https://doi.org/10.1002/chem.201503762, 2016.
Hou, D., Pascazio, L., Martin, J., Zhou, Y., Kraft, M., and You, X.: On the reactive coagulation of incipient soot nanoparticles, J. Aerosol Sci., 159, 105866, https://doi.org/10.1016/j.jaerosci.2021.105866, 2022.
Houston Miller, J., Smyth, K. C., and Mallard, W. G.: Calculations of the dimerization of aromatic hydrocarbons: Implications for soot formation, Symposium (International) on Combustion, 20, 1139–1147, https://doi.org/10.1016/S0082-0784(85)80604-4, 1985.
Humphrey, W., Dalke, A., and Schulten, K.: VMD: visual molecular dynamics, J. Mol. Graphics, 14, 33–38, https://doi.org/10.1016/0263-7855(96)00018-5, 1996.
Iijima, T.: Molecular Structure of Propane, B. Chem. Soc. Jpn., 45, 1291–1294, https://doi.org/10.1246/bcsj.45.1291, 1972.
Indarto, A.: Soot Growth Mechanisms from Polyynes, Environ. Eng. Sci., 26, 1685–1691, https://doi.org/10.1089/ees.2007.0325, 2008.
Irimiea, C., Faccinetto, A., Mercier, X., Ortega, I.-K., Nuns, N., Therssen, E., Desgroux, P., and Focsa, C.: Unveiling trends in soot nucleation and growth: When secondary ion mass spectrometry meets statistical analysis, Carbon, 144, 815–830, https://doi.org/10.1016/j.carbon.2018.12.015, 2019.
Jacobson, R. S., Korte, A. R., Vertes, A., and Miller, J. H.: The Molecular Composition of Soot, Angew. Chem. Int. Edit., 59, 4484–4490, https://doi.org/10.1002/anie.201914115, 2020.
Johansson, K. O., Head-Gordon, M. P., Schrader, P. E., Wilson, K. R., and Michelsen, H. A.: Resonance-stabilized hydrocarbon-radical chain reactions may explain soot inception and growth, Science, 361, 997–1000, https://doi.org/10.1126/science.aat3417, 2018.
Johansson, K. O., Dillstrom, T., Elvati, P., Campbell, M. F., Schrader, P. E., Popolan-Vaida, D. M., Richards-Henderson, N. K., Wilson, K. R., Violi, A., and Michelsen, H. A.: Radical–radical reactions, pyrene nucleation, and incipient soot formation in combustion, P. Combust. Inst., 36, 799–806, https://doi.org/10.1016/j.proci.2016.07.130, 2017.
Kazakov, A. and Frenklach, M.: Dynamic Modeling of Soot Particle Coagulation and Aggregation: Implementation With the Method of Moments and Application to High-Pressure Laminar Premixed Flames, Combust. Flame, 114, 484–501, https://doi.org/10.1016/S0010-2180(97)00322-2, 1998.
Kelesidis, G. A. and Goudeli, E.: Self-preserving size distribution and collision frequency of flame-made nanoparticles in the transition regime, P. Combust. Inst., 38, 1233–1240, https://doi.org/10.1016/j.proci.2020.07.147, 2021.
Kelesidis, G. A., Goudeli, E., and Pratsinis, S. E.: Flame synthesis of functional nanostructured materials and devices: Surface growth and aggregation, P. Combust. Inst., 36, 29–50, https://doi.org/10.1016/j.proci.2016.08.078, 2017a.
Kelesidis, G. A., Goudeli, E., and Pratsinis, S. E.: Morphology and mobility diameter of carbonaceous aerosols during agglomeration and surface growth, Carbon, 121, 527–535, https://doi.org/10.1016/j.carbon.2017.06.004, 2017b.
Kiefer, J. H., Sidhu, S. S., Kern, R. D., Xie, K., Chen, H., and Harding, L. B.: The Homogeneous Pyrolysis of Acetylene II: The High Temperature Radical Chain Mechanism, Combust. Sci. Technol., 82, 101–130, https://doi.org/10.1080/00102209208951815, 1992.
Kim, J. and Ihee, H.: Theoretical study on the reaction of butadiynyl radical (C4H) with ethylene (C2H4) to form C6H4 and H, Int. J. Quantum Chem., 112, 1913–1925, https://doi.org/10.1002/qua.23147, 2012.
Krep, L., Roy, I. S., Kopp, W., Schmalz, F., Huang, C., and Leonhard, K.: Efficient Reaction Space Exploration with ChemTraYzer-TAD, J. Chem. Inf. Model., 62, 890–902, https://doi.org/10.1021/acs.jcim.1c01197, 2022.
Lammertsma, K. and Ohwada, T.: Three-Center, Two-Electron Systems. Origin of the Tilting of Their Substituents, J. Am. Chem. Soc., 118, 7247–7254, https://doi.org/10.1021/ja960004x, 1996.
Langer, R., Mao, Q., and Pitsch, H.: A detailed kinetic model for aromatics formation from small hydrocarbon and gasoline surrogate fuel combustion, Combust. Flame, 258, 112574, https://doi.org/10.1016/j.combustflame.2022.112574, 2023.
Li, P., Zhang, X., and Shi, M.: Recent developments in cyclopropene chemistry, Chem. Commun., 56, 5457–5471, https://doi.org/10.1039/D0CC01612H, 2020.
Lide, D. R. and Christensen, D.: Molecular Structure of Propylene, J. Chem. Phys., 35, 1374–1378, https://doi.org/10.1063/1.1732055, 1961.
Lieske, L.-A., Commodo, M., Martin, J. W., Kaiser, K., Benekou, V., Minutolo, P., D'Anna, A., and Gross, L.: Portraits of Soot Molecules Reveal Pathways to Large Aromatics, Five-/Seven-Membered Rings, and Inception through π-Radical Localization, ACS Nano, 17, 13563–13574, https://doi.org/10.1021/acsnano.3c02194, 2023.
Liu, L., Xu, H., Zhu, Q., Ren, H., and Li, X.: Soot formation of n-decane pyrolysis: A mechanistic view from ReaxFF molecular dynamics simulation, Chem. Phys. Lett., 760, 137983, https://doi.org/10.1016/j.cplett.2020.137983, 2020.
Liu, M., Chu, T.-C., Jocher, A., Smith, M. C., Lengyel, I., and Green, W. H.: Predicting polycyclic aromatic hydrocarbon formation with an automatically generated mechanism for acetylene pyrolysis, Int. J. Chem. Kinet., 53, 27–42, https://doi.org/10.1002/kin.21421, 2021.
Lu, Y.-H., Li, K., and Lu, Y.-W.: Microwave-assisted direct synthesis of butene from high-selectivity methane, Roy. Soc. Open Sci., 4, 171367, https://doi.org/10.1098/rsos.171367, 2017.
Lümmen, N.: Aggregation of carbon in an atmosphere of molecular hydrogen investigated by ReaxFF-molecular dynamics simulations, Comp. Mater. Sci., 49, 243–252, https://doi.org/10.1016/j.commatsci.2010.04.046, 2010.
Mao, Q., van Duin, A. C. T., and Luo, K. H.: Formation of incipient soot particles from polycyclic aromatic hydrocarbons: A ReaxFF molecular dynamics study, Carbon, 121, 380–388, https://doi.org/10.1016/j.carbon.2017.06.009, 2017.
Maricq, M. M.: Coagulation dynamics of fractal-like soot aggregates, J. Aerosol Sci., 38, 141–156, https://doi.org/10.1016/j.jaerosci.2006.11.004, 2007.
Maricq, M. M.: Examining the Relationship Between Black Carbon and Soot in Flames and Engine Exhaust, Aerosol Sci. Tech., 48, 620–629, https://doi.org/10.1080/02786826.2014.904961, 2014.
Martin, J. W., Pascazio, L., Menon, A., Akroyd, J., Kaiser, K., Schulz, F., Commodo, M., D'Anna, A., Gross, L., and Kraft, M.: π-Diradical Aromatic Soot Precursors in Flames, J. Am. Chem. Soc., 143, 12212–12219, https://doi.org/10.1021/jacs.1c05030, 2021.
Mei, J., Wang, M., You, X., and Law, C. K.: Quantitative measurement of particle size distributions of carbonaceous nanoparticles during ethylene pyrolysis in a laminar flow reactor, Combust. Flame, 200, 15–22, https://doi.org/10.1016/j.combustflame.2018.11.010, 2019.
Michelsen, H. A., Colket, M. B., Bengtsson, P.-E., D'Anna, A., Desgroux, P., Haynes, B. S., Miller, J. H., Nathan, G. J., Pitsch, H., and Wang, H.: A Review of Terminology Used to Describe Soot Formation and Evolution under Combustion and Pyrolytic Conditions, ACS Nano, 14, 12470–12490, https://doi.org/10.1021/acsnano.0c06226, 2020.
Miller, J. H.: The kinetics of polynuclear aromatic hydrocarbon agglomeration in flames, Symposium (International) on Combustion, 23, 91–98, https://doi.org/10.1016/S0082-0784(06)80246-8, 1991.
Mukut, K. M., Roy, S., and Goudeli, E.: Molecular arrangement and fringe identification and analysis from molecular dynamics (MAFIA-MD): A tool for analyzing the molecular structures formed during reactive molecular dynamics simulation of hydrocarbons, Comput. Phys. Commun., 276, 108325, https://doi.org/10.1016/j.cpc.2022.108325, 2022 (code available at: https://github.com/comp-comb/MAFIA-MD, last access: February 2025).
Mukut, K. M., Ganguly, A., Goudeli, E., Kelesidis, G. A., and Roy, S. P.: Characterization of Nascent Soot Particles from Acetylene Pyrolysis: A Molecular Modeling Perspective, in: 13th US National Combustion Meeting, Texas, USA, 19–22 March 2023.
Mukut, K. M., Ganguly, A., Goudeli, E., Kelesidis, G. A., and Roy, S. P.: Physical, chemical and morphological evolution of incipient soot obtained from molecular dynamics simulation of acetylene pyrolysis, Fuel, 373, 132197, https://doi.org/10.1016/j.fuel.2024.132197, 2024.
Naseri, A., Kholghy, M. R., Juan, N. A., and Thomson, M. J.: Simulating yield and morphology of carbonaceous nanoparticles during fuel pyrolysis in laminar flow reactors enabled by reactive inception and aromatic adsorption, Combust. Flame, 237, 111721, https://doi.org/10.1016/j.combustflame.2021.111721, 2022.
O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R.: Open Babel: An open chemical toolbox, J. Cheminformatics, 3, 33, https://doi.org/10.1186/1758-2946-3-33, 2011.
Plimpton, S.: Fast Parallel Algorithms for Short-Range Molecular Dynamics, J. Comput. Phys., 117, 1–19, https://doi.org/10.1006/jcph.1995.1039, 1995 (code available at https://www.lammps.org/download.html, last access: March 2024).
Ramanathan, V., Crutzen, P. J., Lelieveld, J., Mitra, A. P., Althausen, D., Anderson, J., Andreae, M. O., Cantrell, W., Cass, G. R., Chung, C. E., Clarke, A. D., Coakley, J. A., Collins, W. D., Conant, W. C., Dulac, F., Heintzenberg, J., Heymsfield, A. J., Holben, B., Howell, S., Hudson, J., Jayaraman, A., Kiehl, J. T., Krishnamurti, T. N., Lubin, D., McFarquhar, G., Novakov, T., Ogren, J. A., Podgorny, I. A., Prather, K., Priestley, K., Prospero, J. M., Quinn, P. K., Rajeev, K., Rasch, P., Rupert, S., Sadourny, R., Satheesh, S. K., Shaw, G. E., Sheridan, P., and Valero, F. P. J.: Indian Ocean Experiment: An integrated analysis of the climate forcing and effects of the great Indo-Asian haze, J. Geophys. Res.-Atmos., 106, 28371–28398, https://doi.org/10.1029/2001JD900133, 2001.
Rissler, J., Messing, M. E., Malik, A. I., Nilsson, P. T., Nordin, E. Z., Bohgard, M., Sanati, M., and Pagels, J. H.: Effective Density Characterization of Soot Agglomerates from Various Sources and Comparison to Aggregation Theory, Aerosol Sci. Tech., 47, 792–805, https://doi.org/10.1080/02786826.2013.791381, 2013.
Rokstad, O. A., Lindvaag, O. A., and Holmen, A.: Acetylene Pyrolysis in Tubular Reactor, Int. J. Chem. Kinet., 46, 104–115, https://doi.org/10.1002/kin.20830, 2014.
Rom, N., Hirshberg, B., Zeiri, Y., Furman, D., Zybin, S. V., Goddard III, W. A., and Kosloff, R.: First-Principles-Based Reaction Kinetics for Decomposition of Hot, Dense Liquid TNT from ReaxFF Multiscale Reactive Dynamics Simulations, J. Phys. Chem. C, 117, 21043–21054, https://doi.org/10.1021/jp404907b, 2013.
Russo, C., Tregrossi, A., and Ciajolo, A.: Dehydrogenation and growth of soot in premixed flames, P. Combust. Inst., 35, 1803–1809, https://doi.org/10.1016/j.proci.2014.05.024, 2015.
Russo, C., Alfè, M., Rouzaud, J.-N., Stanzione, F., Tregrossi, A., and Ciajolo, A.: Probing structures of soot formed in premixed flames of methane, ethylene and benzene, P. Combust. Inst., 34, 1885–1892, https://doi.org/10.1016/j.proci.2012.06.127, 2013.
Sabbah, H., Biennier, L., Klippenstein, S. J., Sims, I. R., and Rowe, B. R.: Exploring the Role of PAHs in the Formation of Soot: Pyrene Dimerization, J. Phys. Chem. Lett., 1, 2962–2967, https://doi.org/10.1021/jz101033t, 2010.
Saggese, C., Sánchez, N. E., Frassoldati, A., Cuoci, A., Faravelli, T., Alzueta, M. U., and Ranzi, E.: Kinetic Modeling Study of Polycyclic Aromatic Hydrocarbons and Soot Formation in Acetylene Pyrolysis, Energ. Fuels, 28, 1489–1501, https://doi.org/10.1021/ef402048q, 2014.
Saxe, P. and Schaefer III, H. F.: Can cyclopropyne really be made?, J. Am. Chem. Soc., 102, 3239-3240, https://doi.org/10.1021/ja00529a057, 1980.
Schmalz, F., Kopp, W. A., Goudeli, E., and Leonhard, K.: Reaction path identification and validation from molecular dynamics simulations of hydrocarbon pyrolysis, Int. J. Chem. Kinet., 56, 501–512, https://doi.org/10.1002/kin.21719, 2024.
Schuetz, C. A. and Frenklach, M.: Nucleation of soot: Molecular dynamics simulations of pyrene dimerization, P. Combust. Inst., 29, 2307–2314, https://doi.org/10.1016/S1540-7489(02)80281-4, 2002.
Schulz, F., Commodo, M., Kaiser, K., De Falco, G., Minutolo, P., Meyer, G., D'Anna, A., and Gross, L.: Insights into incipient soot formation by atomic force microscopy, P. Combust. Inst., 37, 885–892, https://doi.org/10.1016/j.proci.2018.06.100, 2019.
Scienomics SAS: MAPS platform 4.3, http://www.scienomics.com/ (last access: 3 April 2025), 2020.
Sharma, A., Mukut, K. M., Roy, S. P., and Goudeli, E.: The coalescence of incipient soot clusters, Carbon, 180, 215–225, https://doi.org/10.1016/j.carbon.2021.04.065, 2021.
Shiraiwa, M., Ueda, K., Pozzer, A., Lammel, G., Kampf, C. J., Fushimi, A., Enami, S., Arangio, A. M., Fröhlich-Nowoisky, J., Fujitani, Y., Furuyama, A., Lakey, P. S. J., Lelieveld, J., Lucas, K., Morino, Y., Pöschl, U., Takahama, S., Takami, A., Tong, H., Weber, B., Yoshino, A., and Sato, K.: Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 51, 13545–13567, https://doi.org/10.1021/acs.est.7b04417, 2017.
Shukla, B. and Koshi, M.: A novel route for PAH growth in HACA based mechanisms, Combust. Flame, 159, 3589–3596, https://doi.org/10.1016/j.combustflame.2012.08.007, 2012.
Skeen, S. A. and Yasutomi, K.: Measuring the soot onset temperature in high-pressure n-dodecane spray pyrolysis, Combust. Flame, 188, 483–487, https://doi.org/10.1016/j.combustflame.2017.09.030, 2018.
Slavinskaya, N., Mirzayeva, A., Whitside, R., Starke, J., Abbasi, M., Auyelkhankyzy, M., and Chernov, V.: A modelling study of acetylene oxidation and pyrolysis, Combust. Flame, 210, 25–42, https://doi.org/10.1016/j.combustflame.2019.08.024, 2019.
Solà, M.: Aromaticity rules, Nat. Chem., 14, 585–590, https://doi.org/10.1038/s41557-022-00961-w, 2022.
Sun, B., Rigopoulos, S., and Liu, A.: Modelling of soot coalescence and aggregation with a two-population balance equation model and a conservative finite volume method, Combust. Flame, 229, 111382, https://doi.org/10.1016/j.combustflame.2021.02.028, 2021.
Swope, W. C., Andersen, H. C., Berens, P. H., and Wilson, K. R.: A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters, J. Chem. Phys., 76, 637–649, https://doi.org/10.1063/1.442716, 1982.
Tanzawa, T. and Gardiner, W. C.: Reaction mechanism of the homogeneous thermal decomposition of acetylene, J. Phys. Chem., 84, 236–239, https://doi.org/10.1021/j100440a002, 1980.
Thomson, M. J.: Modeling soot formation in flames and reactors: Recent progress and current challenges, P. Combust. Inst., 39, 805–823, https://doi.org/10.1016/j.proci.2022.07.263, 2023.
Wang, Y. and Chung, S. H.: Soot formation in laminar counterflow flames, Prog. Energ. Combust., 74, 152–238, https://doi.org/10.1016/j.pecs.2019.05.003, 2019.
Wang, Y., Gu, M., Liu, D., and Huang, X.: Soot growth mechanism in C2H2 combustion with H2 addition: A reactive molecular dynamics study, Int. J. Hydrogen Energ., 48, 8696–8706, https://doi.org/10.1016/j.ijhydene.2022.12.001, 2022a.
Wang, Y., Gu, M., Wu, J., Cao, L., Lin, Y., and Huang, X.: Formation of soot particles in methane and ethylene combustion: A reactive molecular dynamics study, Int. J. Hydrogen Energ., 46, 36557–36568, https://doi.org/10.1016/j.ijhydene.2021.08.125, 2021.
Wang, Y., Gu, M., Zhu, Y., Cao, L., Wu, J., Lin, Y., and Huang, X.: Analysis of soot formation of CH4 and C2H4 with H2 addition via ReaxFF molecular dynamics and pyrolysis–gas chromatography/mass spectrometry, J. Energy Inst., 100, 177–188, https://doi.org/10.1016/j.joei.2021.11.007, 2022b.
Yuan, H., Kong, W., Liu, F., and Chen, D.: Study on soot nucleation and growth from PAHs and some reactive species at flame temperatures by ReaxFF molecular dynamics, Chem. Eng. Sci., 195, 748–757, https://doi.org/10.1016/j.ces.2018.10.020, 2019.
Zádor, J., Fellows, M. D., and Miller, J. A.: Initiation Reactions in Acetylene Pyrolysis, J. Phys. Chem.-A, 121, 4203–4217, https://doi.org/10.1021/acs.jpca.7b03040, 2017.
Zhang, X., Di, N., Xu, L., Chen, H., Shu, X., Wang, Y., and Lin, Y.: Study on the formation process of soot from 2,5-dimethylfuran pyrolysis by ReaxFF molecular dynamics, J. Therm. Anal. Calorim., 148, 9145–9166, https://doi.org/10.1007/s10973-023-12301-2, 2023.
Zhao, J., Lin, Y., Huang, K., Gu, M., Lu, K., Chen, P., Wang, Y., and Zhu, B.: Study on soot evolution under different hydrogen addition conditions at high temperature by ReaxFF molecular dynamics, Fuel, 262, 116677, https://doi.org/10.1016/j.fuel.2019.116677, 2020.